Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Introduction à la Bio-Informatique
Nadia El-Mabrouk

2
1. Qu’est-ce que la Bio-Informatique?
Champs multi-disciplinaire qui utilise des méthodes informatiques (mathématiques, statistiques, combinatoires…) pour résoudre un problème biologique : Formaliser des problèmes de biologie moléculaire; Développer des outils formels; Analyser les données; Prédire des résultats biologiques; Organiser les données. Discipline relativement nouvelle, qui évolue en fonction des nouveaux problèmes posés par la biologie moléculaire. Pas de consensus sur la définition de la bio-informatique.
 pour résoudre un. problème biologique : Formaliser des problèmes de biologie moléculaire; Développer des outils formels; Analyser les données; Prédire des résultats biologiques; Organiser les données. Discipline relativement nouvelle, qui évolue en fonction des. nouveaux problèmes posés par la biologie moléculaire. Pas de consensus sur la définition de la bio-informatique.")
3
Les séquences d’ADN et de protéines
La Bio-Informatique s’applique à tout type de données biologiques, en particulier moléculaires : Les séquences d’ADN et de protéines Les structures d’ARN et de protéines Les contenus en gènes des génomes Les puces à ADN (microarrays) Les réseaux d’interactions entre protéines Les réseaux métaboliques Les arbres de phylogénie Utilités : Faire avancer les connaissances en biologie, en génétique humaine, en théorie de l’évolution… Aider à la conception de médicaments Comprendre les maladies complexes..
 Les réseaux d’interactions entre protéines. Les réseaux métaboliques. Les arbres de phylogénie. Utilités : Faire avancer les connaissances en biologie, en génétique humaine, en théorie de l’évolution… Aider à la conception de médicaments. Comprendre les maladies complexes..")
4
2. Défis de la biologie moléculaire
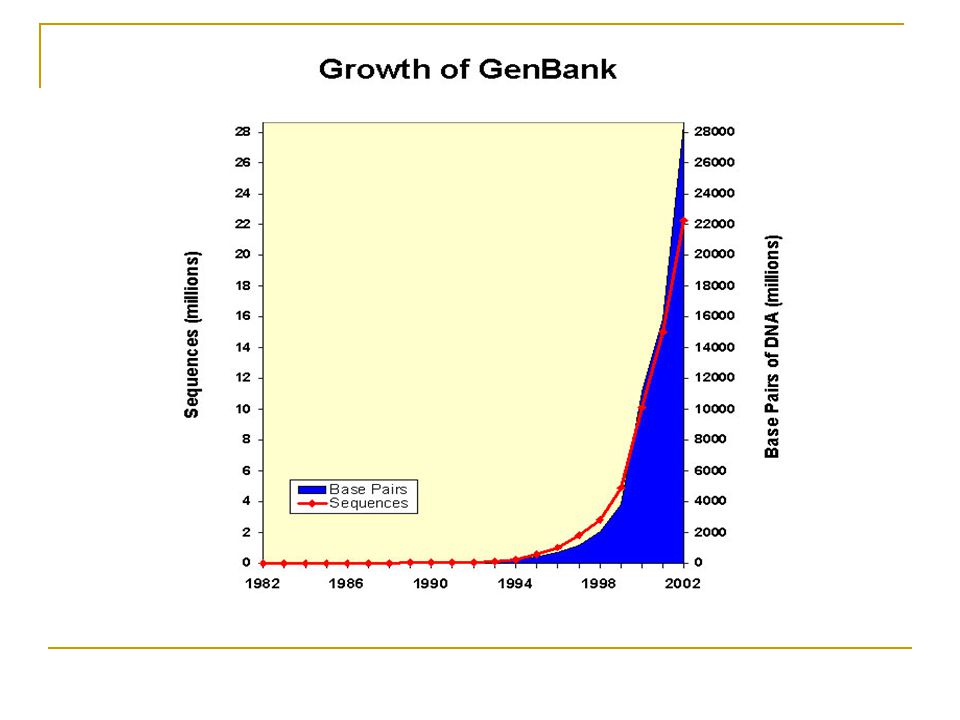
Analyser, comprendre et organiser une masse de données biologiques: Plus de 200 génomes complètement séquencés et publiés, dont l’homme (23 paires de chros.) et la souris (20 paires de chro.) Projet HapMap du génome humain: Construction de la carte des haplotypes Projets de séquençage de plus de 500 procaryotes et 400 eucaryotes
 et la souris (20 paires de chro.) Projet HapMap du génome humain: Construction de la carte des haplotypes. Projets de séquençage de plus de 500 procaryotes et 400 eucaryotes.")
6
Défis de la biologie moléculaire
Décoder l’information contenue dans les séquences d’ADN et de protéines Trouver les gènes Différencier entre introns et exons Analyser les répétitions dans l’ADN Identifier les sites des facteurs de transcription Étudier l’évolution des génomes Génomique structurale: Modéliser les structures 3D des protéines et des ARN structurels Déterminer la relation entre structure et fonction Génomique fonctionnelle Étudier la régulation des gènes Déterminer les réseaux d’interaction entre les protéines

7
3. Les bases de données bioinformatiques les plus utilisées
NCBI, National Center for Biotechnology Information GenBank: Séquences d’ADN (3 billion de paires de bases) Site officiel de BLAST PubMed: Permet la recherche de références COGs: Familles de gènes orthologues … EMBL, The European Molecular Biology Laboratory ExPASy, Expert Protein Analysis System, Protéomique Swiss-Prot: Séquences de protéines PROSITE: Domaines et familles de protéines SWISS-MODEL: Outil de prédiction 3D de protéines Différents outils de recherche PDB, Protein Data Bank Base de données de structures 3D de protéines Visualisation et manipulation de structures SCOP, Structural Classification of Proteins
 Site officiel de BLAST. PubMed: Permet la recherche de références. COGs: Familles de gènes orthologues … EMBL, The European Molecular Biology Laboratory. ExPASy, Expert Protein Analysis System, Protéomique. Swiss-Prot: Séquences de protéines. PROSITE: Domaines et familles de protéines. SWISS-MODEL: Outil de prédiction 3D de protéines. Différents outils de recherche. PDB, Protein Data Bank. Base de données de structures 3D de protéines. Visualisation et manipulation de structures. SCOP, Structural Classification of Proteins.")
8
4. Intérêt des séquences La séquence nucléotidique d’un gène détermine la séquence d’acides aminés de la protéine La séquence d’une protéine détermine sa structure et sa fonction Généralement, une similarité de séquence implique une similarité de structure et de fonction (l’inverse n’est pas toujours vrai) Évolution basée, en partie, sur la duplication suivie de modification (« bricolage évolutif »). D’où, beaucoup de redondance dans les bases de données
 Évolution basée, en partie, sur la duplication suivie de modification (« bricolage évolutif »). D’où, beaucoup de redondance dans les bases de données.")
9
4.1 Recherche dans les bases de données
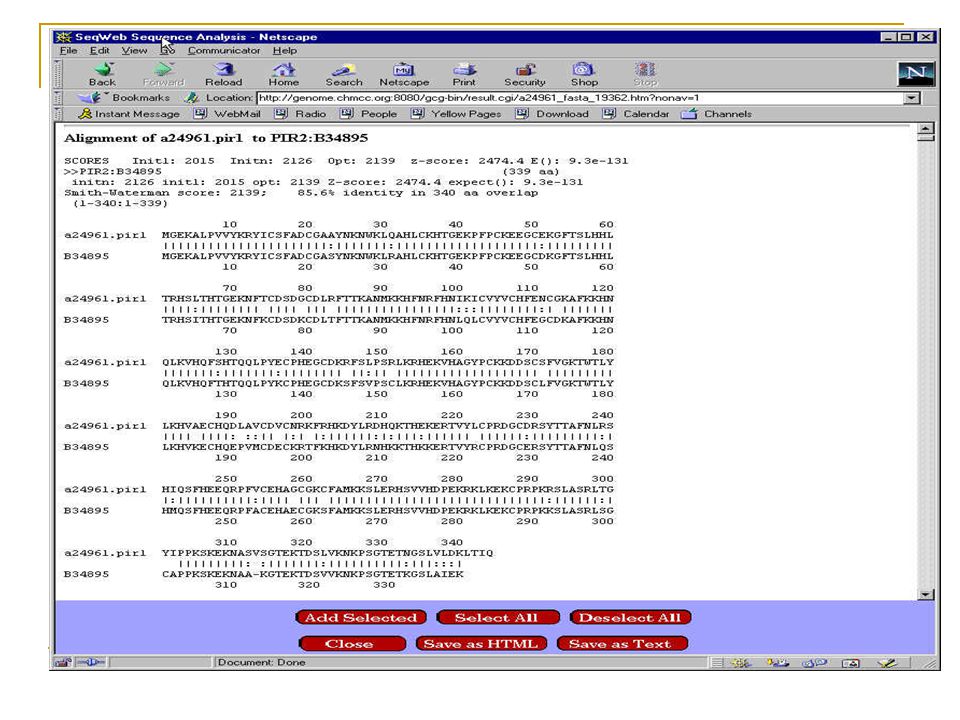
Tache courante d’un biologiste moléculaire Est-ce qu’une nouvelle séquence a déjà été complètement ou partiellement déposée dans les bases de données? Est-ce que cette séquence contient un gène? Est-ce que ce gène appartient à une famille connue? Quelle est la protéine encodée? Existe-t-il d’autres gènes homologues? Existe-t-il des séquences non-codantes similaires. Répétitions ou séquences régulatrices Logiciels les plus connus: Smith-Waterman, FASTA et BLAST

10
4.2 Alignement local et global
Alignement de deux séquences: Méthodes naturelle pour comparer deux séquences. On compte le nombre de ``différences’’ (insertion, suppression, substitution) Alignement Global: C A G C A – C G T G G A T T C T C G G | | | | | | | | | | | T A T C A G C G T G G – C A C T A G C Alignement Local: CAGCAC T T – G G A T TCTCGG | | | | | TAGT T T A G G - T GGCAT Recherche: C A G C A – C T T G G A T T C T C G G | | | | | | C A G C G T G G
 Alignement Global: C A G C A – C G T G G A T T C T C G G. | | | | | | | | | | | T A T C A G C G T G G – C A C T A G C. Alignement Local: CAGCAC T T – G G A T TCTCGG. | | | | | TAGT T T A G G - T GGCAT. Recherche: C A G C A – C T T G G A T T C T C G G. | | | | | | C A G C G T G G.")
11
Signification de l’alignement de séquences
Modèle sous-jacent: Mutations ponctuelles Exemple: Substitution de caractère Séquence ancestrale inconnue G C G | | A C G ACG A B Séquences observées A G GCG ACG

13
Comparaison de deux génomes

14
4.3 Alignement multiple Trouver des caractéristiques communes à une famille de protéines Relier la séquence à la structure et à la fonction Caractériser les gènes homologues Caractériser les régions conservées et les régions variables Déduire des contraintes de structures pour les ARN Construire des arbres de phylogénie

16
Leishmaniose Leishmania (Kinetoplastida) Phlebotomus (Diptera)
Sinclair Stammers/TDR/OMS Phlebotomus (Diptera)
")
17
Phlébotomes Plus de 800 espèces différentes 1-3 millimètres

18
Comment reconnaitre un phlébotome
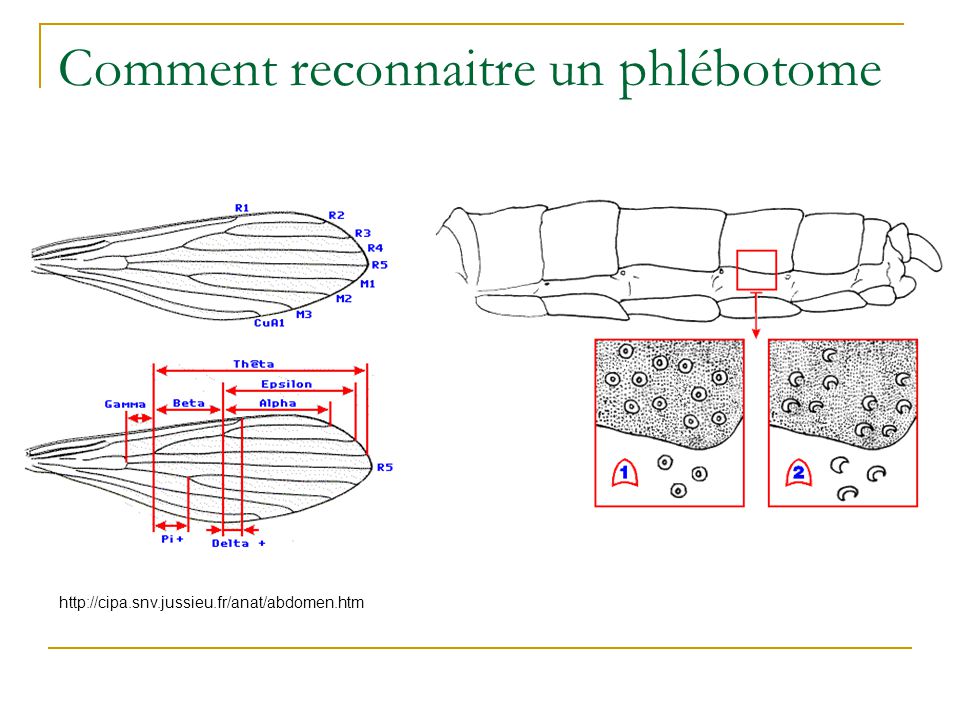
19
CIPA (Computer-aided Identification of Phlebotomine sandlies of America

20
CIPA (Computer-aided Identification of Phlebotomine sandlies of America

21
CIPA (Computer-aided Identification of Phlebotomine sandlies of America

22
CIPA (Computer-aided Identification of Phlebotomine sandlies of America

23
La biodiversité

24
Classification naturelle = phylogénie

25
Combien existe-t-il d’arbres ?
3 espèces : 3 arbres D A B C 4 espèces : 5 * 3 arbres n espèces : (2n-3)(2n – 5) (2n – 7) … (3) (1) arbres 10 espèces : espèces :
(2n – 5) (2n – 7) … (3) (1) arbres. 10 espèces : espèces :")
26
Arbres de phylogénie Racine: Ancêtre commun
Feuilles: Espèces actuelles Nœuds internes: Points de spéciation Taille des branches: Temps d’évolution

27
Types de données et Méthodes
Séquences d’ADN ou de protéines Présence/absence ou Ordre des gènes Méthodes Alignement de séquence Calcul de distances Minimisation du nombre de mutations Approches probabilistes de maximum de vraisemblance

28
Développement de l'Oursin Paracentrotus lividus

29
Réseau de régulation

30
Développement précoce du mésoderme d’oursin
[Copyright: H. Bolouri & E. Davidson, < (2001)]
]")
31
Modélisation Une partie importante de la bioinformatique est la modélisation de systèmes complexes, comme les réseaux de régulations. Le but est d’avoir un système un peu moins compliqué dans le but de pourvoir l’analyser et possiblement prédire des phénomènes de régulation. MAIS COMMENT CHOISIR NOTRE MODÈLE: Buts Modèle Données

32
Modèles détaillés versus …
Un modèle détaillé avec beaucoup de paramètres Peut représenter des phénomènes très précis du réseau - la concentration des protéines - les réactions cinétiques Par contre, demande un nombre très grand de données pour l’analyse du modèle et l’inférence de résultats

33
… modèles grossiers Un modèle grossier avec peu de paramètres
Représenter des phénomènes grossiers du réseau - exemple: un gène est « on » ou « off » Requiert un petit nombre de données pour l’analyse du réseau Par contre, les résultats inférés peuvent être très loin de la réalité

34
Modèles discrets versus…
Un modèle discret représente le réseau à un moment précis dans le temps Exemple: réseau booléen sommet : gène est « on » = 1 ou « off » = 0 arête : interaction entre deux gènes deux états: présente ou absente On peut ensuite modéliser les influences positives ou négatives des différents gènes par des fonctions booléennes Avantage: simplicité Inconvénient: trop restrictif -> réseau booléen probabiliste ??

35
… modèles continus Un modèle continu représente le réseau à travers le temps Dans ce cas, le réseau est modélisé par un système d’équations différentielles Les variables du système sont les concentrations à travers le temps Avantage : système représentant la réalité Inconvénient : dimension du système qui croît trop vite

Présentations similaires




>")
>")













 Parcours Ingénierie de lIntelligence.>")
