Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Obésité. Poids de la génétique.
L. Metzinger I. Définition clinique – Situation mondiale. Ia. L’épidémie d’obésité dans le monde. Prévalence. Ib. Situation en France. Evolution et prévalence. II. Hérédité. IIa. Poids de l’hérédité génétique. Les expériences sur jumeaux monozygotes. IIc. Les gènes humains impliqués dans l’obésité monogénique. Syndromes de Prader-Willi et de Barbet-Biedl. IIb. Les études multigéniques. Dernières avancées sur la recherche des gènes de susceptibilité à l’obésité. III. Les gènes humains impliqués dans l’obésité monogénique. IIIa. La leptine et son récepteur. IIIb. Le neuropeptide Y et son récepteur. IIIc. La protéine Agouti et le récepteur à la mélanocortine MC4R. IIId. Récepteur 3-adrénergique. IIIe. Protéines découplantes UCP-2 et UCP-3. IIIf. Peroxysome proliferator activated receptors (PPAR). IV. Perspectives. Apports de la nutrigénétique.
. IV. Perspectives. Apports de la nutrigénétique.")
2
Carte de France. Prévalence de l’obésité chez l’adulte par région en 2000 (Institut Roche de l'Obésité, 2000). : Prévalence du surpoids et de l’obésité dans la population masculine française par tranche d’âge en 2000

3
Evolution de l’esthétique à travers les ages. La beauté est subjective.. .
Les faibles consommateurs de nourriture devaient vraisemblablement mieux survivre en période de famine que les individus gaspilleurs de calories. Mais en période d'abondance, ce rendement métabolique performant aboutit à un stockage excessif de graisses de réserve.

4
Quantification : indice de masse corporelle IMC body mass index (BMI)
Poids en kg/ (taille en m)2 Si IMC entre 18.5 et 25, norme. Si IMC entre 25 et 30, surpoids Si IMC entre 30 et 35, obésité modérée Si IMC entre 35 et 40, obésité sévère Si IMC supérieur à 40, obésité morbide ou massive
2. Si IMC entre 18.5 et 25, norme. Si IMC entre 25 et 30, surpoids. Si IMC entre 30 et 35, obésité modérée. Si IMC entre 35 et 40, obésité sévère. Si IMC supérieur à 40, obésité morbide ou massive.")
6
Obésité androide vs obésité gynoide
. Type androïde : prédominance à la partie supérieure du corps (taille, thorax) avec un rapport tour de taille sur tour de hanches >1 ; Type gynoïde : prédominance au niveau du bassin, des hanches avec un rapport taille sur hanche < 0,8 ; Type mixte.
 avec un rapport tour de taille sur tour de hanches >1 ; Type gynoïde : prédominance au niveau du bassin, des hanches avec un rapport taille sur hanche < 0,8 ; Type mixte.")
7
Conséquences secondaires de l’obésité morbide
Hypertension artérielle Hypercholésterolémie Diabète de type 2 L’apnée du sommeil Syndrome de l'hypoventilation liée à l'obésité Brûlures d'estomac et reflux gastrique Asthme et bronchite Calculs de la vésicule biliaire. Incontinence à l'effort Altération discale dégénérative Arthrose des articulations portantes Stase veineuse Perturbations affectives et psychologiques Conséquences néfastes sur le plan social

8
Obésité et Génétique Syndromes monogéniques :
Prader-Willi (1/ ) Obésité (>200% poids idéal), petite taille, hypogonadisme, retard mental, désordres mentaux liés à la recherche de nourriture, comportement aggrissif, automutilation. Tetée difficille à la naissance. Fertilité diminuée. Locus 15q Gène candidat : SMN (small nuclear ribonucleoprotein-associatd polypeptide). Délétion chez 70% des patients. Régulation de l ’épissage des ARNm dans le cerveau. Barbet-Biedl (1/17500, autosomique récessif). Obésité, hypogénitalisme masculin, retard mental dystrophie rétinale, anomalie rénale (50% diabète type II), Hypertension (50%). Maladie hétérogène : mutations sur chromosome 3, 15 et 16. Leptine et récepteur à la leptine.
 Obésité (>200% poids idéal), petite taille, hypogonadisme, retard mental, désordres mentaux liés à la recherche de nourriture, comportement aggrissif, automutilation. Tetée difficille à la naissance. Fertilité diminuée. Locus 15q Gène candidat : SMN (small nuclear ribonucleoprotein-associatd polypeptide). Délétion chez 70% des patients. Régulation de l ’épissage des ARNm dans le cerveau. Barbet-Biedl (1/17500, autosomique récessif). Obésité, hypogénitalisme masculin, retard mental dystrophie rétinale, anomalie rénale (50% diabète type II), Hypertension (50%). Maladie hétérogène : mutations sur chromosome 3, 15 et 16. Leptine et récepteur à la leptine.")
9
Obésité et Génétique Syndromes multigéniques : Modèle non-mendéllien. Contribution héréditaire dans l ’obésité commune complexe, interaction cumulative de plusieurs gènes ayant chacun un faible effet individuel = gènes de susceptibilité . L ’obésité massive familiale serait influencée par quelques gènes majeurs. Jumeaux monozygotes (Bouchard et al., New Engl. J. Med., 1990) Paires de jumeaux males hétérozygotes, régime hypercalorique : 90 % de variation entre paires, 10 % inter-paires (Masse graisseuse) Différences éthniques : indiens américains Pima (70 % obésité et résistance à l ’insuline, 40 % diabètes) trait de survie pour la sélection génétique avant l ’arrivée de la société de consommation.
 Paires de jumeaux males hétérozygotes, régime hypercalorique : 90 % de variation entre paires, 10 % inter-paires (Masse graisseuse) Différences éthniques : indiens américains Pima (70 % obésité et résistance à l ’insuline, 40 % diabètes) trait de survie pour la sélection génétique avant l ’arrivée de la société de consommation.")
10
Hérédité de l’obésité (Bouchard C.)
5% 25% Génétique 30% 30% Génétique Culturelle Culturelle 65% 45% Non transmissible Non transmissible % de graisse corporelle Masse graisseuse Indice de Masse corporelle

11
Gènes candidats : comment les trouver ?
Cartographie des maladies multifactorielles. Modèle murin : Il existe à ce jour au moins 20 loci répartis sur le génome associés à certains critères d ’obésité. Les modèles animaux multigéniques reproduisant spontanément les caractéristiques de l ’obésité humaine sont essentiels à l ’étude des anomalies génétiques responsables de la maladie. Homogénéité génétique, contrôle de l ’environnement, sélection des croisements et donc simplification de la recherche et de l ’identification des gènes candidats. Stratégie : analyse de la liaison génétique pour rechercher la co-transmission de certains génotypes avec le phénotype représentant la maladie. Pour cela, on suit des marqueurs.

12
Gènes candidats : comment les trouver ?
Cartographie des maladies multifactorielles. RFLP (Restriction Fragment Length Polymorphism) Ce sont les premiers polymorphismes mis en évidence (1978). Ils sont révélés par des modifications de la carte de restriction d’un ADN donné, autrement dit de la façon dont une enzyme de restriction va couper l’ADN génomique d’un individu. Ceci est classiquement mis en évidence grâce à la méthode de Southern. Les minisatellites : Ce sont des séquences répétitives, dispersées sur tout le génome et très polymorphes. Il s’agit d’une famille de séquences répétées ayant en commun un motif central de 11 à 16 pb. Les minisatellites sont dispersés dans tout le génome humain, dans des endroits bien précis sur la carte humaine qu’on appelle régions hypervariables. Elle se présentent sous la forme d’une répétition successive de n fois le motif central (de 10 à plusieurs centaines de fois). Le polymorphisme réside dans le nombre de répétitions des motifs centraux. Microsatellites ou STR (Short Tandem Repeats). Il s’agit là encore d’une répétition successive de n motifs identiques, la différence étant que le motif est très court : 2 à 4 nucléotides.
 Ce sont les premiers polymorphismes mis en évidence (1978). Ils sont révélés par des modifications de la carte de restriction d’un ADN donné, autrement dit de la façon dont une enzyme de restriction va couper l’ADN génomique d’un individu. Ceci est classiquement mis en évidence grâce à la méthode de Southern. Les minisatellites : Ce sont des séquences répétitives, dispersées sur tout le génome et très polymorphes. Il s’agit d’une famille de séquences répétées ayant en commun un motif central de 11 à 16 pb. Les minisatellites sont dispersés dans tout le génome humain, dans des endroits bien précis sur la carte humaine qu’on appelle régions hypervariables. Elle se présentent sous la forme d’une répétition successive de n fois le motif central (de 10 à plusieurs centaines de fois). Le polymorphisme réside dans le nombre de répétitions des motifs centraux. Microsatellites ou STR (Short Tandem Repeats). Il s’agit là encore d’une répétition successive de n motifs identiques, la différence étant que le motif est très court : 2 à 4 nucléotides.")
13
Gènes candidats : comment les trouver ?
Cartographie des maladies multifactorielles. Le bût est d ’étudier une population de marqueurs répartis sur tout le génome, afin d ’isoler des marqueurs, qui dans une situation idéale, seront systématiquement présents chez les souris malades et systématiquement absents chez les souris saines. Les gènes impliqués seront proches physiquement du ou des marqueurs trouvés. Dans les maladies multigéniques, on prouvera plutôt que certains marqueurs se retrouvent plus souvent chez les malades que chez les sains (lod score). Chez l ’humain, la stratégie est la même, mais les croisements ne sont pas contrôlés par le chercheur :-) . On travaille sur des cohortes de familles de malades avec pédigré (et donc méïose informative).
. Chez l ’humain, la stratégie est la même, mais les croisements ne sont pas contrôlés par le chercheur :-) . On travaille sur des cohortes de familles de malades avec pédigré (et donc méïose informative).")
15
Nature 387, (1997); doi: /43185 Congenital leptin deficiency is associated with severe early-onset obesity in humans CARL T. MONTAGUE*†, I. SADAF FAROOQI*†‡, JONATHAN P. WHITEHEAD*‡, MARIA A. SOOS*‡, HARALD RAU*‡, NICHOLAS J. WAREHAM§, CIARAN P. SEWTER*‡, JANET E. DIGBY*‡, SHEHLA N. MOHAMMED, JANE A. HURST¶, CHRISTOPHER H. CHEETHAM&Num;#, ALISON R. EARLEY&Num;#, ANTHONY H. BARNETT, JOHANNES B. PRINS*‡ & STEPHEN O&Apos;RAHILLY* Figure 1 Detection of a homozygous frame-shift mutation in the leptin genes of probands Ob1 and Ob2. Adipocyte RNA was isolated from a needle biopsy of subcutaneous adipose tissue obtained from Ob2. RNA was reverse-transcribed and the nucleotide sequence encoding the leptin protein was amplified by PCR and subcloned, and four subclones were sequenced. The nucleotide sequence provided by the automated sequencing apparatus corresponds to the reverse complement of the coding sequence. In all four clones of Ob2's leptin cDNA, five, rather than the expected six, guanine nucleotides were present between nucleotides 393 and 398. The same region of the leptin coding sequence was amplified from genomic DNA of Ob1 and an unrelated control subject. The leptin gene of the control subject has six guanines (G) in the appropriate position (shown as cysteines (C), as the reverse complement strand was sequenced), whereas Ob1 is homozygous for the deleted guanine.
 in the appropriate position (shown as cysteines (C), as the reverse complement strand was sequenced), whereas Ob1 is homozygous for the deleted guanine.")
16
Leptine et récepteur à la leptine : des souris et des hommes.
Obésité extrême détectée chez cinq membres de deux familles qui sont mutants homozygotes du gène codant la leptine (décalage du cadre de lecture chez une famille, mutation faux-sens chez l ’autre). Même cause et même pathologie que la souris ob/ob. Obésité extrême détectée chez trois membres de la même famille qui sont mutants homozygotes du gène codant le récepteur de la leptine (erreur d ’épissage). Même cause et même pathologie que la souris db/db. Obésité modérée chez les gens mutants hétérozygotes du gène codant la leptine (une copie normale, une copie mutée).
. Même cause et même pathologie que la souris ob/ob. Obésité extrême détectée chez trois membres de la même famille qui sont mutants homozygotes du gène codant le récepteur de la leptine (erreur d ’épissage). Même cause et même pathologie que la souris db/db. Obésité modérée chez les gens mutants hétérozygotes du gène codant la leptine (une copie normale, une copie mutée).")
17
A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction Karine Clément, Christian Vaisse, Najiba Lahlou, Sylvie Cabrol, Veronique Pelloux, Dominique Cassuto, Micheline Gourmelen, Christian Dina, Jean Chambaz, Jean-Marc Lacorte, Arnaud Basdevant, Pierre Bougn?res, Yves Lebouc, Philippe Froguel, Bernard Guy-Grand Nature392, (26 Mar 1998) Letters to Nature Figure 2 SSCP scanning and DNA- sequence analysis of exon 16 of the human leptin receptor. a, An homozygous mobility shift in exon 16 was seen for the proband HD416 as compared with two control obese subjects (C1 and C2) unrelated to the family. Here mother, HD409, was heterozygous for the same variant conformer. b, Sequence analysis (reverse complementary strand shown) of the exon 16/intron 16 junction of HD416 and of a control subject. There is a G A base substitution in the splice donor site of exon 16 in patient HD416.
 Letters to Nature. Figure 2 SSCP scanning and DNA- sequence analysis of exon 16 of the human leptin receptor. a, An homozygous mobility shift in exon 16 was seen for the proband HD416 as compared with two control obese subjects (C1 and C2) unrelated to the family. Here mother, HD409, was heterozygous for the same variant conformer. b, Sequence analysis (reverse complementary strand shown) of the exon 16/intron 16 junction of HD416 and of a control subject. There is a G A base substitution in the splice donor site of exon 16 in patient HD416.")
18
Effet du traitement par la leptine (2 ans) chez les enfants mutés
 chez les enfants mutés")
19
Boucle régulatrice leptine-Neuropeptide Y
La leptine et l’insuline passent la barrière hematoméningée, pour signaler aux neurones des noyaux arqués la quantité d’énergie stockée sous formes de lipides. Les neurones exprimant le neuropeptide Y (NPY) et agouti-related protein (AgRP) stimulent la prise de nourriture et diminuent la dépense d’énergie, donc augmentation pondérale (stimulation de l’anabolisme). L’expression de la proopiomelanocortin (POMC) et de son dérivé alpha-MSH ainsi que de cocaine- and amphetamine-regulated transcript (CART) inhibe la prise de nourriture et augmente la dépense d’énergie, donc diminution pondérale (stimulation du catabolisme). De haut niveaux de leptine et d’insuline inhibent les neurones AgRP/NPY et stimulent les neurones POMC/CART tandis que des niveaux bas induisent l’inverse.
 et agouti-related protein (AgRP) stimulent la prise de nourriture et diminuent la dépense d’énergie, donc augmentation pondérale (stimulation de l’anabolisme). L’expression de la proopiomelanocortin (POMC) et de son dérivé alpha-MSH ainsi que de cocaine- and amphetamine-regulated transcript (CART) inhibe la prise de nourriture et augmente la dépense d’énergie, donc diminution pondérale (stimulation du catabolisme). De haut niveaux de leptine et d’insuline inhibent les neurones AgRP/NPY et stimulent les neurones POMC/CART tandis que des niveaux bas induisent l’inverse.")
20
ObRb : activation du systeme JAK-STAT

21
Signalisation intracellulaire : OBRb

22
Gènes candidats, cibles thérapeutiques ?
NPY et récepteur au NPY : NPY stimule la prise de nourriture, mais aussi contrôle de l ’humeur, excitabilité cérébrocorticale, signalisation hypothalamus-glande pituitaire, physiologie cardiovasculaire et fonction sympathique. Récepteur dans l ’hypothalamus. Souris KO pour NPY : prise de nourriture et poids normal, mais hyperphagie lorsque mise à jeun. Souris Ko pour NPYR1 : mangent moins que les contrôles mais diminution de l’activité et du métabolisme basal => obèses. Alpha MSH (melanocyte stimulating hormone) et récepteur MC4R. Antagoniste : Agouti related protein. Souris KO pour aMSH et autres dérivés POMC ont un phénotype obésité, insuffisance surrénalienne, pigmentation altérée. Il existe des humains ayant un défaut génétique et des symptômes similaires. Souris KO pour MC4R : surcharge pondérale, mangent plus que les contrôles, dépensent moins d’énergie. Augmentation de la masse graisseuse. Chez les humains, mutation du MC4R expliquerait 4% des obésités avec IMC > 35. Injection intracérébrale d’un analogue aMSH chez le rat diminue la prise de nourriture.
 et récepteur MC4R. Antagoniste : Agouti related protein. Souris KO pour aMSH et autres dérivés POMC ont un phénotype obésité, insuffisance surrénalienne, pigmentation altérée. Il existe des humains ayant un défaut génétique et des symptômes similaires. Souris KO pour MC4R : surcharge pondérale, mangent plus que les contrôles, dépensent moins d’énergie. Augmentation de la masse graisseuse. Chez les humains, mutation du MC4R expliquerait 4% des obésités avec IMC > 35. Injection intracérébrale d’un analogue aMSH chez le rat diminue la prise de nourriture.")
23
GAD 2. GAD2 code la glutamate décarboxylase (production de l'acide gamma-aminobutyrique ou GABA) - neurones NPY contiennent du GABA - l'hormone gastrique PYY 3-36 agit en inhibant les neurones à neuropeptide Y et en désinhibant les neurones répondant à la leptine par un mécanisme qui fait intervenir le GABA gène GAD2 dans une région de liaison génétique à l'obésité au locus l0p12 gène candidat de l'obésité. L'activité du gène GAG2 est multipliée par 6 chez les porteurs de l'allèle G-243, variant retrouvé en excès dans les populations d'obèses massifs.

24
Gènes candidats, cibles thérapeutiques ?
Protéines découplantes de type 2 et 3 (UCP2 et 3). Les UCP (uncoupling proteins) découplent le gradient de protons produit par l ’oxydation des aliments et l ’énergie potentielle créée par ce gradient n ’est pas convertie en ATP mais perdue sous forme de chaleur. UCP2 ubiquitaire, UCP3 muscle squelettique. Souris surexprimant UCP3 : hyperphages et maigres. Souris KO pour UCP3 : phénotype normal (?!?) Plusieurs polymorphismes du gène codant UCP3 liés avec prédisposition à gagner de la masse graisseuse. Synergie entre mutations de UCP3 et récepteur adrénergique 3 ?
. Les UCP (uncoupling proteins) découplent le gradient de protons produit par l ’oxydation des aliments et l ’énergie potentielle créée par ce gradient n ’est pas convertie en ATP mais perdue sous forme de chaleur. UCP2 ubiquitaire, UCP3 muscle squelettique. Souris surexprimant UCP3 : hyperphages et maigres. Souris KO pour UCP3 : phénotype normal ( ! ) Plusieurs polymorphismes du gène codant UCP3 liés avec prédisposition à gagner de la masse graisseuse. Synergie entre mutations de UCP3 et récepteur adrénergique 3")
25
Gènes candidats, cibles thérapeutiques ?
Récepteur adrénergique 3 : Exprimé dans adipocytes, règle lipolyse des triglycérides (adipocytes blancs) et thérmogénèse (adipocytes bruns). Sept domaines transmembranaires, couplage à protéine Gs. Indiens PIMA, mutation faux-sens Try64-Arg associée à obésité et début de diabète non-insulino-dépendant chez homozygotes. Retrouvée chez deux fois plus d ’obèses japonais que de non obèses. Résultats non retrouvés en Europe. Souris KO : prise de nourriture et poids normal, augmentation modeste des réserves graisseuses, surtout chez les femelles. Essai clinique avec agonistes 3 : quelques effets sur dépense d’énergie mais peu de résultats probants en adminsitration chronique.
 et thérmogénèse (adipocytes bruns). Sept domaines transmembranaires, couplage à protéine Gs. Indiens PIMA, mutation faux-sens Try64-Arg associée à obésité et début de diabète non-insulino-dépendant chez homozygotes. Retrouvée chez deux fois plus d ’obèses japonais que de non obèses. Résultats non retrouvés en Europe. Souris KO : prise de nourriture et poids normal, augmentation modeste des réserves graisseuses, surtout chez les femelles. Essai clinique avec agonistes 3 : quelques effets sur dépense d’énergie mais peu de résultats probants en adminsitration chronique.")
26
Différenciation adipocytaire : gènes candidats, cibles thérapeutiques ?
L ’accroissement de la masse adipeuse peut provenir soit d ’une augmentation de la taille des adipocytes (hypertophie, obésité légère), soit de leur nombre (hyperplasie, obésité sévère), soit des deux. Deux types de gènes sont impliqués dans le processus de différenciation des cellules adipeuses et pourraient altérer le processus. PPAR- (peroxysome proliferator activated receptor) Famille des récepteurs hormonaux nucléaires. Deux isoformes. Exprimé fortement dans les adipocytes. Activés par clofibrate et dérivés d ’acides gras. Transfection de PPAR- 2 dans fibroblastes avec activateurs conversion en adipocytes.
, soit de leur nombre. (hyperplasie, obésité sévère), soit des deux. Deux types de gènes sont impliqués dans le processus de différenciation des cellules adipeuses et pourraient altérer le processus. PPAR- (peroxysome proliferator activated receptor) Famille des récepteurs hormonaux nucléaires. Deux isoformes. Exprimé fortement dans les adipocytes. Activés par clofibrate et dérivés d ’acides gras. Transfection de PPAR- 2 dans fibroblastes avec activateurs conversion en adipocytes.")
27
PPAR gamma : des actions multiples

28
Ligands des PPAR PPAR: PPAR: PPAR:
Acides gras polyinsaturés et dérivés oxydés Cible des fibrates (hypocholestérolémiants) Phénotype obèse chez souris KO PPAR: Acides gras polyinsaturés Prostaglandines PPAR: Dérivés des acides gras: acides hydroxyoctadécadiénoïques (HODE) Prostaglandines: 15-désoxy12,14-prostaglandine J2 Cible des Glitazones (insulino-sensibilisants) Souris KO +/- : 15% de masse adipeuse en moins. Traitement avec antagonistes : diminution taille adipocytes, insuline, glucose, masse graisseuse.
 Phénotype obèse chez souris KO. PPAR: Acides gras polyinsaturés. Prostaglandines. PPAR: Dérivés des acides gras: acides hydroxyoctadécadiénoïques (HODE) Prostaglandines: 15-désoxy12,14-prostaglandine J2. Cible des Glitazones (insulino-sensibilisants) Souris KO +/- : 15% de masse adipeuse en moins. Traitement avec antagonistes : diminution taille adipocytes, insuline, glucose, masse graisseuse.")
29
Actions des PPAR Foie PPAR HDL Transport inverse
du cholestérol Oxydation des ac. gras Synthèse des VLDL Lipolyse Triglycérides Petites LDL denses HDL

30
Actions des PPAR Muscles, adipocytes, macrophages PPAR HDL
Transport inverse du cholestérol Oxydation des ac. gras Masse corporelle Sensibilité à l’insuline HDL

31
Actions des PPAR Adipocytes PPAR Muscles PPAR Métabolisme des
ac. gras libres Cytokines Sensibilité à l’insuline Homéostasie glucidique Fonction vasculaire Muscles PPAR

32
Différenciation adipocytaire : gènes candidats, cibles thérapeutiques ?
C/EBP- et (CCAAT/enhancer binding protein) Famille des leucine-zipper. Expression plus tardives que PPAR- . Exprimé fortement dans les adipocytes. Synergie entre C/EBP et PPAR- : co-transfection dans fibroblastes sans activateurs très forte conversion en adipocytes. Elément de reconnaissance pour C/EBP dans le promoteur du gène codant la leptine ADD1 (adipocyte determination and differentiation-dependant factor1)/SERBP protéine de type hélice-boucle-hélice exprimée très tôt dans l ’adipogénèse. Augmente l ’expression de PPAR- Contrôle l ’expression de protéines clefs dans l ’homéostasie du cholestérol.
 Famille des leucine-zipper. Expression plus tardives que PPAR- . Exprimé fortement dans les adipocytes. Synergie entre C/EBP et PPAR- : co-transfection dans fibroblastes sans activateurs. très forte conversion en adipocytes. Elément de reconnaissance pour C/EBP dans le promoteur du gène codant la leptine. ADD1 (adipocyte determination and differentiation-dependant factor1)/SERBP. protéine de type hélice-boucle-hélice exprimée très tôt dans l ’adipogénèse. Augmente l ’expression de PPAR- Contrôle l ’expression de protéines clefs dans l ’homéostasie du cholestérol.")
33
Nutrigénétique Etude des interactions gènes-environnement et de la variabilité de la réponse d'un individu Puces à ADN (microarray) : analyse simultanée des étapes clés de l’homéostasie cellulaire. diagnostic Evaluation de nouvelles molécules, d’alicaments et de régimes spécifiques.
 : analyse simultanée des étapes clés de l’homéostasie cellulaire. diagnostic. Evaluation de nouvelles molécules, d’alicaments et de régimes spécifiques.")
34
2007 : l’équipe américano-britannique du Professeur Andrew Hattersley (Exeter, GB) et l’équipe franco-anglo-suisse du Professeur Philippe Froguel variante du gène FTO chez les personnes souffrant d’obésité. Les personnes qui possèdent une copie de la mutation du gène FTO (fatso ou fat mass and obesity associated gene) ont 30% plus de risques de devenir obèse que les autres. Et celles qui possèdent deux copies du gène FTO modifié risquent à 70% de devenir obèse. 16% d'adultes ont deux copies de la variante du gène FTO, pèsent en moyenne 3 kilos de plus que ceux qui ne l'ont pas. Ces personnes présente un risque d'obésité de 67% plus élevé que les autres. l'association entre les SNP du gène FTO et l'IMC a augmenté les risques de l'obésité dans la communauté Amish. Les gènes FTO associés à la prise de poids et l'obésité ont été relevés dans 30% de la population européenne, ce qui représente 3 kilos de masse corporelle en plus. la masse de données recueillie est suffisante pour montrer que "La prise de poids résultant de la présence de cette prédisposition génétique est beaucoup plus faible et statistiquement insignifiante chez les sujets physiquement très actifs (3-4 h activité physique modérée/jour).
 ont 30% plus de risques de devenir obèse que les autres. Et celles qui possèdent deux copies du gène FTO modifié risquent à 70% de devenir obèse. 16% d adultes ont deux copies de la variante du gène FTO, pèsent en moyenne 3 kilos de plus que ceux qui ne l ont pas. Ces personnes présente un risque d obésité de 67% plus élevé que les autres. l association entre les SNP du gène FTO et l IMC a augmenté les risques de l obésité dans la communauté Amish. Les gènes FTO associés à la prise de poids et l obésité ont été relevés dans 30% de la population européenne, ce qui représente 3 kilos de masse corporelle en plus. la masse de données recueillie est suffisante pour montrer que La prise de poids résultant de la présence de cette prédisposition génétique est beaucoup plus faible et statistiquement insignifiante chez les sujets physiquement très actifs (3-4 h activité physique modérée/jour).")
35
Trois nouveaux gènes associés à l'obésité et à la prise de poids sont identifiés Les chercheurs ont aussi trouvé des variations de l'ADN à proximité des gènes MAF et PTER (18 Janvier 2009 Nature Genetics équipe de Philippe Froguel et David Meyre ), et directement dans la séquence codante du gène NPC1. Ces polymorphismes génétiques, largement répandus dans les populations européennes, modulent le risque d'obésité sévère et la prise de poids en population générale tout au long de la vie. Le gène NPC1 comporte plus de 200 mutations pathogéniques responsables de la maladie de Niemann-Pick type C, une atteinte neurodégénérative progressive. Les souris privées de NPC1 présentent, en plus de désordres neurologiques, une perte de poids et un manque d'appétit. La mutation associée à l'obésité pourrait donc induire une augmentation de la fonction de la protéine NPC1, celle-ci fonctionnant trop bien dès lors que le gène NPC1 est muté. Le gène MAF, lui, code pour une protéine particulière qui est impliquée dans la différenciation du tissu adipeux (tissu en charge du stockage des graisses) et dans la production et la sécrétion de l'insuline et du glucagon. Un dernier gène (PRL) est associé à l'obésité et à la prise de poids plus spécifiquement chez l'adulte. PRL produit la prolactine, une hormone bien connue pour son effet déclencheur de la lactation chez la femme. La prolactine joue aussi un rôle dans le contrôle de la prise alimentaire.
 et dans la production et la sécrétion de l insuline et du glucagon. Un dernier gène (PRL) est associé à l obésité et à la prise de poids plus spécifiquement chez l adulte. PRL produit la prolactine, une hormone bien connue pour son effet déclencheur de la lactation chez la femme. La prolactine joue aussi un rôle dans le contrôle de la prise alimentaire..")
36
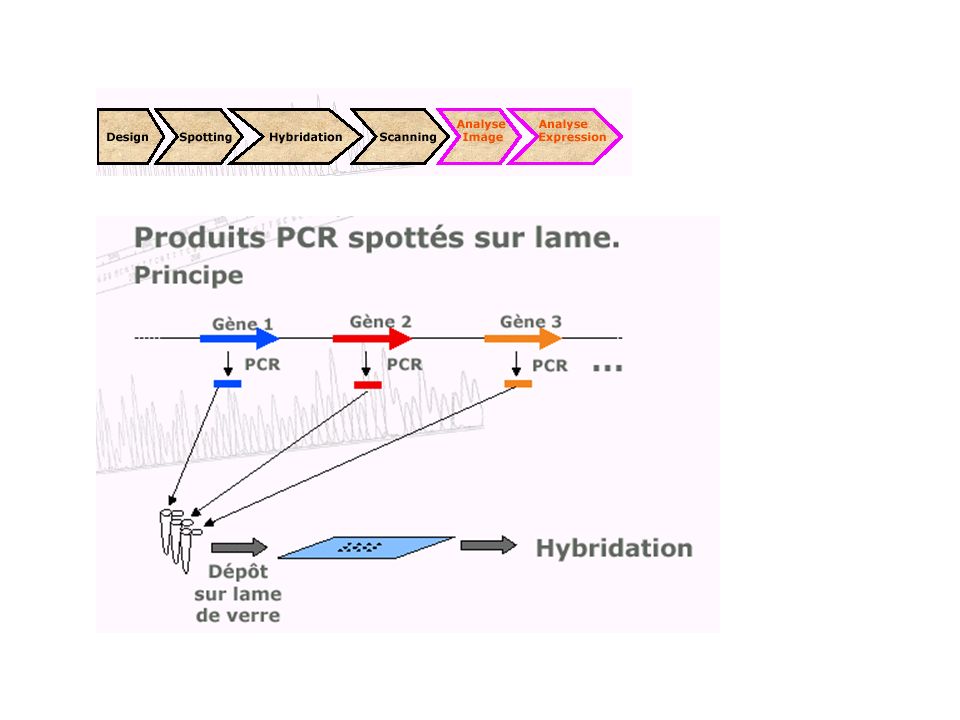
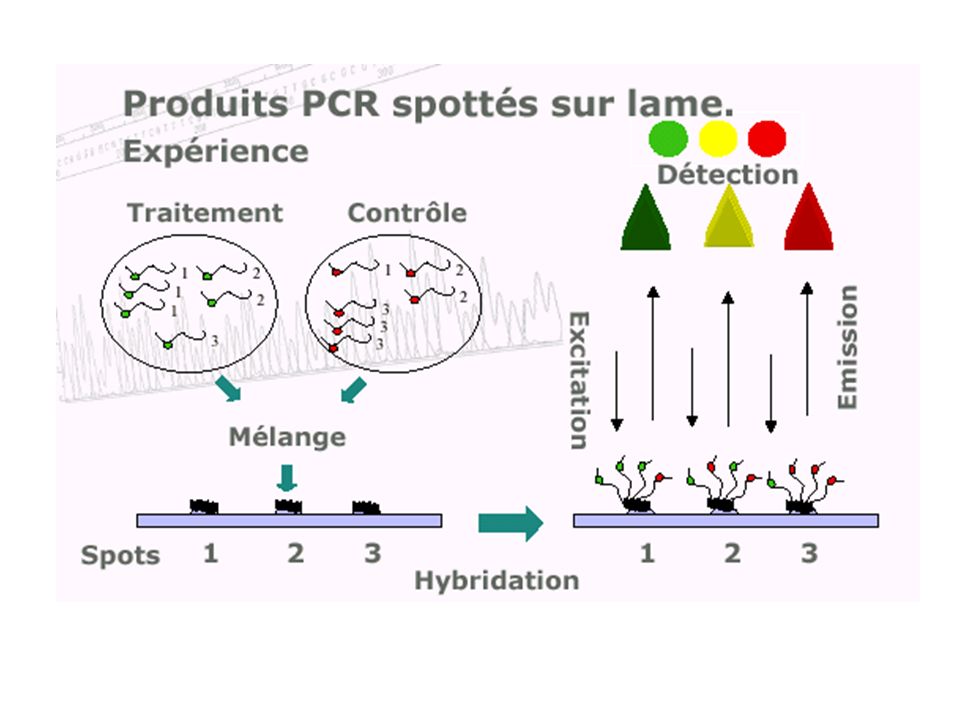
Transcriptome : suivre l ’expression
caractériser et quantifier les produits d'expression de l'ADN (les ARN messagers) de manière à identifier, dans un tissu, dans un état et à un moment donné du développement, les séquences actives et donc à révéler le niveau d ’expression des gènes dont elles sont issues. Avant : Northern blotting RT-PCR Ce qui reste : la sonde (probe) et la cible (target) Ce qui change : la TGV ! (high input…)
 de manière à identifier, dans un tissu, dans un état et à un moment donné du développement, les séquences actives et donc à révéler le niveau d ’expression des gènes dont elles sont issues. Avant : Northern blotting. RT-PCR. Ce qui reste : la sonde (probe) et la cible (target) Ce qui change : la TGV ! (high input…)")
42
Les lésions de l ’ADN humain. Classement.
A. Perturbations chromosomiques : excès ou perte de chromosome ou d'une partie d'un chromosome. Peut impliquer tous les gènes présents dans la région touchée. Ex. Syndrome de Down (trisomie 21). B. Maladies monogéniques : un seul gène muté. Subdivisé en : autosomique dominant (un allèle touché suffit) - autosomique récessif (les 2 allèles doivent être mutés) - liée au sexe : chr. X (chez l'homme, un seul X. Si muté, maladie. Chez la femme, dépend si trait dominant ou récessif).
. B. Maladies monogéniques : un seul gène muté. Subdivisé en : - autosomique dominant (un allèle touché suffit) - autosomique récessif (les 2 allèles doivent être mutés) - liée au sexe : chr. X (chez l homme, un seul X. Si muté, maladie. Chez la femme, dépend si trait dominant ou récessif).")
43
Les lésions de l ’ADN humain. Classement.
C. Maladies polygéniques, multifactorielles : Situation beaucoup plus compliquée. Ne suit pas la génétique Mendélienne classique. Sont classés dedans l'hypertension, l'obésité, le diabète, de nombreuses maladies cardio-vasculaires, désordres psychiatriques etc... Trait complexe : n'importe quel phénotype dont l'hérédité n'est pas mendélienne, donc n'est pas attribuable à un seul gène. Autres termes s'appliquant à ces maladies : * maladies multifactorielles : interaction entre l'environnement, l'hérédité culturelle (famille...) et l'hérédité génétique. * polygénique : interaction de plusieurs gènes. Par suite du polymorphisme dans ces gènes, les gens prédisposés à la maladie vont avoir un ensemble de plusieurs gènes interdépendants qui vont fonctionner, mais moins bien que la moyenne. Ces gènes vont interagir les uns avec les autres et par effet cascade, et en fonction de l'environnement entraîner la maladie.
 et l hérédité génétique. * polygénique : interaction de plusieurs gènes. Par suite du polymorphisme dans ces gènes, les gens prédisposés à la maladie vont avoir un ensemble de plusieurs gènes interdépendants qui vont fonctionner, mais moins bien que la moyenne. Ces gènes vont interagir les uns avec les autres et par effet cascade, et en fonction de l environnement entraîner la maladie.")
44
Quelques rappels ADN ARN Protéine
Appariements Watson-Crick : A-T et G-C ADN 5 ’ 3 ’ Promoteur Exons + introns 5 ’ 3 ’ TRANSCRIPTION + Epissage ARN 5 ’ AUG UAA UAG UGA AAAAA 3 ’ Exons 5 ’NTR 3 ’NTR TRADUCTION Protéine NH2 COOH

45
Les lésions de l ’ADN humain
I. Macrolésions : Délétion : excision d’un segment d’ADN avec rétablissement de la continuité de la double hélice. Insertion : introduction d’un segment d’ADN avec rétablissement de la continuité de la double hélice. Inversion : changement d’orientation, en tête-bêche, d’un segment plus ou moins long d’ADN. Duplication : duplication d’une partie d’ADN, la copie s’insérant à la suite du modèle. 5 ’ 3 ’ 5 ’ 3 ’ 5 ’ 3 ’ 5 ’ 3’ 5 ’ 3’ 5 ’ 3 ’ 3 ’ 5’ 3’ 5 ’ 5 ’ 3’ 3’ 5’ 3’ 5’ 5’ 3’ 5 ’ 3’ 5 ’ 3’ 3 ’ 5’ 3 ’ 5’

46
Les lésions de l ’ADN humain
II. Microlésions : Substitution. Synonyme GAG (glu) GAA (glu) Faux-sens GAG (glu) GTG (val) Non-sens CAG (gln) TAG (stop) Délétion ou Insertion. Décalage du cadre de lecture. AAG AGT ATC ACT AAG lys ser ile thr lys AAG GAG TAT CAC TAA G AAG AGT ATC --TAA G lys ser ile thr STOP lys glu tyr stop + 1 nt - 2 nt
 GAA (glu) Faux-sens GAG (glu) GTG (val) Non-sens CAG (gln) TAG (stop) Délétion ou Insertion. Décalage du cadre de lecture. AAG AGT ATC ACT AAG. lys ser ile thr lys. AAG GAG TAT CAC TAA G AAG AGT ATC --TAA G. lys ser ile thr STOP lys glu tyr stop. + 1 nt. - 2 nt.")
47
Les lésions de l ’ADN humain
III. Lésions dans des séquences non codantes : Région 5’ NTR et 3’ NTR. Promoteur (TATA box) transcription nulle Eléments de régulation (CAAT Box, CpG islands…) transcription affectée Queue Poly A stabilité affectée Sites d’épissage. Régle de Breathnach et Chambon pour les introns : GT AG Expansion de triplets. Syndrome du X fragile : répétition de triplets dans un intron du gène codant la protéine FMR-1. Séquence codante normale chez les malades, mais présence dans un intron d'une répétition de triplets, CGG. Chez les sujets normaux, CGG est répété 5-54 fois. Dans la 2ème génération, fois. 3ème génération : >200 fois maladie apparaît..
 transcription nulle. Eléments de régulation (CAAT Box, CpG islands…) transcription affectée. Queue Poly A stabilité affectée. Sites d’épissage. Régle de Breathnach et Chambon pour les introns : GT AG. Expansion de triplets. Syndrome du X fragile : répétition de triplets dans un intron du gène codant la protéine FMR-1. Séquence codante normale chez les malades, mais présence dans un intron d une répétition de triplets, CGG. Chez les sujets normaux, CGG est répété 5-54 fois. Dans la 2ème génération, fois. 3ème génération : >200 fois maladie apparaît..")
48
Diagnostic I. ADN Le diagnostic repose sur l’utilisation de l’hybridation spécifique avec des sondes (Southern) ou des amorces (PCR). Analyse de la séquence codante si le gène est cloné : le diagnostic direct. Il met en évidence la lésion génétique elle-même. Pour cela, il faut que le gène soit connu, et que les lésions décrites ne soient pas trop variées. On compare toujours par rapport à un sujet témoin présentant un gène normal. Southern blot. Grosses délétions (100 pb à plusieurs milliers). Apparition ou délétion d’un site de restriction. Nécessite l ’utilisation de composés radioactifs (32P, 35S). Apparition de kits utilisant la chemiluminescence ou des réactions colorimétriques. PCR. Grosses et petites délétions (2 pb à 3000 pb). Nécessite l ’utilisation de bromure d ’éthidium. SSCP. Mutations ponctuelles changeant la structure d’au moins un des brins de l ’ADN muté.
 ou des amorces (PCR). Analyse de la séquence codante si le gène est cloné : le diagnostic direct. Il met en évidence la lésion génétique elle-même. Pour cela, il faut que le gène soit connu, et que les lésions décrites ne soient pas trop variées. On compare toujours par rapport à un sujet témoin présentant un gène normal. Southern blot. Grosses délétions (100 pb à plusieurs milliers). Apparition ou délétion d’un site de restriction. Nécessite l ’utilisation de composés radioactifs (32P, 35S). Apparition de kits utilisant la chemiluminescence ou des réactions colorimétriques. PCR. Grosses et petites délétions (2 pb à 3000 pb). Nécessite l ’utilisation de bromure d ’éthidium. SSCP. Mutations ponctuelles changeant la structure d’au moins un des brins de l ’ADN muté.")
49
Purification d ’ADN génomique
Les leucocytes sont d’abord traité par un détergent et par l’enzyme protéinase K. Ceci permet de libérer l’ADN nucléaire dans le milieu et de digérer les protéines qui lui sont associées. L’ADN est ensuite purifié par une suite d’extractions et de précipitations. La première extraction se fait par le phénol. Les protéines sont solubles dans la phase phénolique alors que les acides nucléiques restent dans la phase aqueuse. Il suffit donc d’ajouter le phénol dans la solution, d’agiter vigoureusement, de centrifuger, puis de prélever la phase aqueuse qui contient les acides nucléiques débarrassés des protéines. La deuxième extraction se fait au chloroforme. Le bût, cette fois-ci, est d’éliminer les traces de phénol contaminant la phase aqueuse.

50
Purification d ’ADN génomique
On précipite ensuite les acides nucléiques. Cette précipitation se fait à haute force ionique en rajoutant de l’éthanol à forte concentration (2.5 vol). Dans ces conditions, la presque totalité de l’ADN est précipitée. Si on a affaire à de petites quantités d’ADN, il est souvent nécessaire de précipiter à froid (4 à -20°C) pour accélérer le processus. Le précipité est ensuite lavé plusieurs fois avec de l’éthanol à 70% pour ôter les traces de sel. Après centrifugation et élimination du surnageant, on pourra récupérer l’ADN sous forme solide et le resolubiliser dans le tampon voulu à la concentration voulue. Le dosage de l’ADN se fait par photométrie, les bases puriques et pyrimidiques absorbant fortement dans l’ultraviolet à 260 nm. Une unité de densité optique à 260 nm correspond à : une solution d’ADN double brin à 50 g/ml ou à une solution d’ARN ou d’ADN simple brin à 40 g/ml.
. Dans ces conditions, la presque totalité de l’ADN est précipitée. Si on a affaire à de petites quantités d’ADN, il est souvent nécessaire de précipiter à froid (4 à -20°C) pour accélérer le processus. Le précipité est ensuite lavé plusieurs fois avec de l’éthanol à 70% pour ôter les traces de sel. Après centrifugation et élimination du surnageant, on pourra récupérer l’ADN sous forme solide et le resolubiliser dans le tampon voulu à la concentration voulue. Le dosage de l’ADN se fait par photométrie, les bases puriques et pyrimidiques absorbant fortement dans l’ultraviolet à 260 nm. Une unité de densité optique à 260 nm correspond à : une solution d’ADN double brin à 50 g/ml ou à. une solution d’ARN ou d’ADN simple brin à 40 g/ml.")
51
Southern , Western et Northern blotting

52
Southern blotting

53
Techniques de blotting

54
Hybridation des amorces
PCR 5 ’ 3 ’ 95°C Dénaturation Hybridation des amorces 5 ’ 3 ’ 3 ’ 5 ’ 5 ’ 3 ’ Elongation

Présentations similaires









>")


 Pr E. Tournier-Lasserve>")

 Pr E. Tournier-Lasserve>")

 polymorphes (entre individus, espèces, …) permettant - l’établissement de cartes.>")







