Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Preformulation et formulation pharmaceutique

2
I- Introduction La pharmacie galénique science de la transformation d’une molécule active en un Medt de qualité qui soit sûr, efficace et adapté à chaque catégorie de patients. La conception d’un nouveau Medt : processus long ,compliquée et couteux Dans l'industrie pharmaceutique, ces processus peuvent-être subdivisés et répartis en quatre phases: La phase de recherche La phase de développement ou conception (Préformulation et formulation…) La phase clinique La phase de mise sur le marché .
 La phase clinique. La phase de mise sur le marché. .")
4
II. Etapes de développement d’un médicament
II.1- Phase de recherche de nouvelles molécules actives II.1.1 Principales voies permettent d’obtenir des molécules II.1.2- les essais précliniques

5
II-2-La phase de conception
II-2-1-Préformulation II-2-2- Formulation II.2.3- Optimisation de la formulation II.2.4- Production industrielle

6
II-2-1- La préformulation:
Définition: l’étude des caractéristiques physico-chimiques, technologiques et biologiques du principe actif nécessaire pour le formuler et développer une forme pharmaceutique stable ayant la biodisponibilité maximale ,tout en étant compatible avec une production industrielle .

7
II-2-1- La préformulation
II Caractéristiques du principe actif a)-Caractères organoleptiques: b)-Caractères physico-chimiques c)-Propriétés technologiques d)-Etude de stabilité - Les différents types d’essais de stabilité - Méthodologie de l’étude de stabilité pour un nouveau PA : - Méthodologie du choix de l’excipient
-Caractères organoleptiques: b)-Caractères physico-chimiques c)-Propriétés technologiques d)-Etude de stabilité - Les différents types d’essais de stabilité - Méthodologie de l’étude de stabilité pour un nouveau PA : - Méthodologie du choix de l’excipient")
8
b-Caractéristiques physico-chimiques
Caractères organoleptiques Aspect, forme, couleur, odeur et goût : l’odeur et le goût détermine l’acceptabilité de la substance (PA amer : formulation en sirop ou gélule). La pureté: La présence d’impuretés peut affecter la stabilité et l’aspect du produit fini. Certaines impuretés sont toxiques Etat physique : état cristallin/amorphe La forme amorphe est plus soluble que les cristaux, mais elle est toujours instable Le PA présente des propriétés pharmacologiques, une stabilité chimique et des comportements technologiques différents selon l’état physique. Ce phénomène peut être mis à profit pour augmenter ou diminuer la vitesse de dissolution et l’absorption d’une substance.
. La pureté: La présence d’impuretés peut affecter la stabilité et l’aspect du produit fini. Certaines impuretés sont toxiques. Etat physique : état cristallin/amorphe. La forme amorphe est plus soluble que les cristaux, mais elle est toujours instable. Le PA présente des propriétés pharmacologiques, une stabilité chimique et des comportements technologiques différents selon l’état physique. Ce phénomène peut être mis à profit pour augmenter ou diminuer la vitesse de dissolution et l’absorption d’une substance.")
9
Source « fournisseur 1 » : Une grande quantité de cristaux de taille moyenne et de forme losange a été observée. Des cristaux de forme rectangulaire en moindre quantité ont été également identifiés (Fig.9). Source « fournisseur 1 » : Une grande quantité de cristaux de taille moyenne et de forme losange a été observée. Des cristaux de forme rectangulaire en moindre quantité ont été également identifiés Photographie obtenue par microscopie électronique sur le A0358 du « fournisseur 1 »

10
Source « fournisseur 2 » : Une grande quantité de gros cristaux de forme trapézoïdale est observée
Photographie obtenue par microscopie électronique sur le A0358 du « fournisseur 2 »

11
Source « fournisseur 3 » : Cette matière présente des cristaux de différentes tailles et des amas de particules de différentes tailles. Les contours de ces particules sont plutôt irréguliers Photographie obtenue par microscopie électronique sur le A0358 du « fournisseur 3 »

12
Polymorphisme : Le polymorphisme est un phénomène par lequel un principe actif peut cristalliser en plusieurs systèmes cristallins distincts où les arrangements moléculaires sont différents de sorte que deux polymorphes d’une même substance diffèrent physiquement (point de fusion, solubilité…). Pour une température et une pression donnée, une seule forme cristalline est stable. Les autres appelées métastables sont généralement plus solubles. Pour des substances dont on connaît l’existence de ce phénomène, il faudra s’assurer que la conservation n’entraîne pas de variation de la forme cristalline.
. Pour une température et une pression donnée, une seule forme cristalline est stable. Les autres appelées métastables sont généralement plus solubles. Pour des substances dont on connaît l’existence de ce phénomène, il faudra s’assurer que la conservation n’entraîne pas de variation de la forme cristalline.")
13
Le pseudopolymorphisme :
Propriété d’une molécule à cristalliser (passer à l’état solide) avec une quantité stoechiométrique de son solvant de cristallisation. Pendant la cristallisation, l’eau et les molécules de solvant peuvent se combiner au principe actif dans des liaisons plus ou moins stables en donnant des solvates et si le milieu est aqueux des hydrates. Les propriétés physiques de ces produits peuvent être très différentes de celle de la forme anhydre.
 avec une quantité stoechiométrique de son solvant de cristallisation. Pendant la cristallisation, l’eau et les molécules de solvant peuvent se combiner au principe actif dans des liaisons plus ou moins stables en donnant des solvates et si le milieu est aqueux des hydrates. Les propriétés physiques de ces produits peuvent être très différentes de celle de la forme anhydre.")
14
Le polymorphisme et le pseudo-polymorphisme peuvent avoir des répercussions importantes :
modification propriétés technologiques (hygroscopicité, stabilité, comprimabilité) et biopharmaceutiques (vitesse de dissolution, absorption) des substances médicamenteuses transformation polymorphique au cours de la granulation, broyage, compression ou lors de la conservation du PA et excipient. Il est donc nécessaire de tenir compte de ce phénomène à fin de pouvoir choisir correctement la forme définitive du PA et surtout assurer sa reproduction. Le Norvir® (médt contre le Sida, laboratoires Abbott) : retiré du marché en 1998 car sa fabrication produisait une nouvelle forme cristalline, qui se révélait être inefficace contre la maladie.
 et biopharmaceutiques (vitesse de dissolution, absorption) des substances médicamenteuses. transformation polymorphique au cours de la granulation, broyage, compression ou lors de la conservation du PA et excipient. Il est donc nécessaire de tenir compte de ce phénomène à fin de pouvoir choisir correctement la forme définitive du PA et surtout assurer sa reproduction. Le Norvir® (médt contre le Sida, laboratoires Abbott) : retiré du marché en 1998 car sa fabrication produisait une nouvelle forme cristalline, qui se révélait être inefficace contre la maladie.")
15
Densité relative:La densité influe sur la stabilité et l’homogénéité du mélange
Point de fusion: Important à connaître, car il détermine la stabilité des molécules à la température. (lors du séchage ou de stérilisation). Solubilité : :le Nb de partie en volume du liquide nécessaire pour dissoudre une partie en masse de la substance considérée. La solubilité dépend : -soluté :nature chimique, granulométrie, état physique. -Le solvant -pH et T° -Si la solubilité est< 10 mg/ml, modifier l’état physique substance ou utiliser artifice de formulation pour la solubilité.
. Solubilité : :le Nb de partie en volume du liquide nécessaire pour dissoudre une partie en masse de la substance considérée. La solubilité dépend : -soluté :nature chimique, granulométrie, état physique. -Le solvant. -pH et T° -Si la solubilité est< 10 mg/ml, modifier l’état physique substance ou utiliser artifice de formulation pour la solubilité.")
16
Formation ester: dissolution retardée de PA
Solubilité par : Formation sel: C’est la méthode de choix pour augmenter la solubilité de certains principes actifs. Formation ester: dissolution retardée de PA Éviter une dégradation au niveau gastrique. Retarder ou prolonger l’action de certains principes actifs. Masquer une saveur désagréable : ester de chloramphénicol. Utilisation de tensio-actif : pouvoir mouillant tension interfaciale S/L qui favorise l’interaction solvant/soluté. Ex: Fungizone : solubilisation par deoxycholate de Na qui forme une micelle dans laquelle l’amphotericine est solubilisé.

17
pKa et Coefficient de partage
Le pKa =pH pr lequel un Ac se présente à 50% S/ forme non ionisée et 50% S/ forme ionisée. Le rapport forme ionisée/non ionisée est défini par les équations d’Henderson-Hasselbach selon le pH du 1/2 ,le rapport fraction ionisée / fraction non ionisée du médt varie. La solubilité d’un acide ou d’une base varie beaucoup avec le pH. La forme ionisée est généralement beaucoup plus soluble dans l’eau donc moins liposoluble. Les acides et les bases sous forme non ionisée sont plus liposolubles. Ac faible : - pH alcalin :ionisation importante, ce qui limitera le passage transMb de cette substance, - pH acide : ionisation faible,, le médt passera mieux les Mb cellulaires. Base faible : on observe l’inverse Exemple : L’acide acétyl salicylique à pH 1,5 existe à 99% sous forme non ionisée, liposoluble capable de ce fait de traverser la muqueuse gastrique.

20
Une classification est proposée pour les conditions suivantes : 25°C, 80% d’HR ambiante et 24 heures de stockage. Classes Comportement très hygroscopique accroissement de la teneur en eau > 15 % hygroscopique accroissement de la teneur en eau de 2 à 5 % légèrement hygroscopique accroissement de la teneur en eau de 0,2 à 2 % non hygroscopique accroissement de la teneur en eau < 0,2 %

21
c- Propriétés technologiques
Influence directe sur l’optimisation de la formulation. Ce sont : C-1- Étude de la distribution granulométrique. C-2- Analyse de surface spécifique. C-3- Critères de comprimabilité C-3-a-Mesure de l’indice de coulabilité C-3-b-Mesure de l’aptitude au tassement. C-3-c- La comprimabilité ou La cohésivité C-3-d- La compressibilité

22
C-1-Etude distribution granulométrique
La granulométrie correspond à la distribution de taille des particules. la forme et l'état de surface des particules : Des particules de petite taille et de forme sphérique auront un comportement favorable à l’écoulement dans des conditions de température et d’humidité définies. les formes éloignées de la sphéricité et la rugosité sont un frein à l'écoulement en favorisant l'enchevêtrement des particules.

24
On constate « d’après l’étude d’écoulement d’une poudre à travers un orifice » un maximum d’écoulement pour une taille optimale qui dépend de la nature de la matière et quand la taille particulaire descend en dessous d’une valeur critique, l’écoulement libre s’annule à cause des forces d’interaction particulaires qui l’emportent sur les forces d’attraction dues à la pesanteur.

25
La mouillabilité θ= 0° : mouillage parfait θ < 90° : mouillage imparfait θ >90° : mouillage nul L'angle de contact θ est défini comme l'angle formé à la jonction des trois phases (solide - liquide - gaz). dépend valeur tension superficielle liquide mouillage ; le mouillage est d’autant + parfait que cette valeur est faible. Toute subs capable de diminuer la tension superficielle favorise le mouillage ex : le pouvoir mouillant des tensio-actifs : diminue la tension interfaciale S/L ce qui favorisera l’interaction solvant (eau)/soluté (PA).
. dépend valeur tension superficielle liquide mouillage ; le mouillage est d’autant + parfait que cette valeur est faible. Toute subs capable de diminuer la tension superficielle favorise le mouillage ex : le pouvoir mouillant des tensio-actifs : diminue la tension interfaciale S/L ce qui favorisera l’interaction solvant (eau)/soluté (PA).")
26
C-2- Surface spécifique
La surface spécifique (ou aire massique) d’une poudre est la surface développée par gramme de produit. Elle est déterminée par la mesure de l’interface solide-gaz, rapportée à l’unité de masse de l’échantillon mesuré. la surface spécifique totale, mesurée par des méthodes d’adsorption gazeuse, prenant en compte la totalité de la surface développée (surface des pores, rugosité des particules,...).
 d’une poudre est la surface développée par gramme de produit. Elle est déterminée par la mesure de l’interface solide-gaz, rapportée à l’unité de masse de l’échantillon mesuré. la surface spécifique totale, mesurée par des méthodes d’adsorption gazeuse, prenant en compte la totalité de la surface développée (surface des pores, rugosité des particules,...).")
27
C-3- Critères de comprimabilité
C Fluidité ou coulabilité de la poudre Un écoulement libre, suffisant de la substance non seulement dans la trémie, le sabot distributeur mais aussi dans la chambre de compression. indispensable à l'obtention de comprimés de masse et de résistance mécanique constantes, quelle que soit la cadence de production. Elle garantit également la constance de la dose thérapeutique. La mesure de l’écoulement de la poudre et l’angle de repos sont important dans le choix de l’excipient et l’optimisation de la performance de la poudre

28
Essai d’écoulement : aptitude des poudres à s'écouler verticalement.
Le test consiste à chronométrer le temps de passage (t) de 100 g de poudre non tassé à travers un entonnoir normalisé. l'essai est réalisé 3 fois. Le temps d’écoulement doit être inférieur à 10 secondes Ce test permet donc d’optimiser la quantité de régulateur d’écoulement qu’il convient d’ajouter. Un agent d’écoulement (lubrifiant) est ajouté aux poudres pour un T >10 sec afin d’améliorer l’écoulement et la fluidité.
 de 100 g de poudre non tassé à travers un entonnoir normalisé. l essai est réalisé 3 fois. Le temps d’écoulement doit être inférieur à 10 secondes. Ce test permet donc d’optimiser la quantité de régulateur d’écoulement qu’il convient d’ajouter. Un agent d’écoulement (lubrifiant) est ajouté aux poudres pour un T >10 sec afin d’améliorer l’écoulement et la fluidité.")
29
Figure : Configuration de l'entonnoir normalisé

30
Angle de repos : mesurer l’angle formé par un tas de poudre dans des conditions d’écoulement données

31
C-3-b-Mesure de l’aptitude au tassement
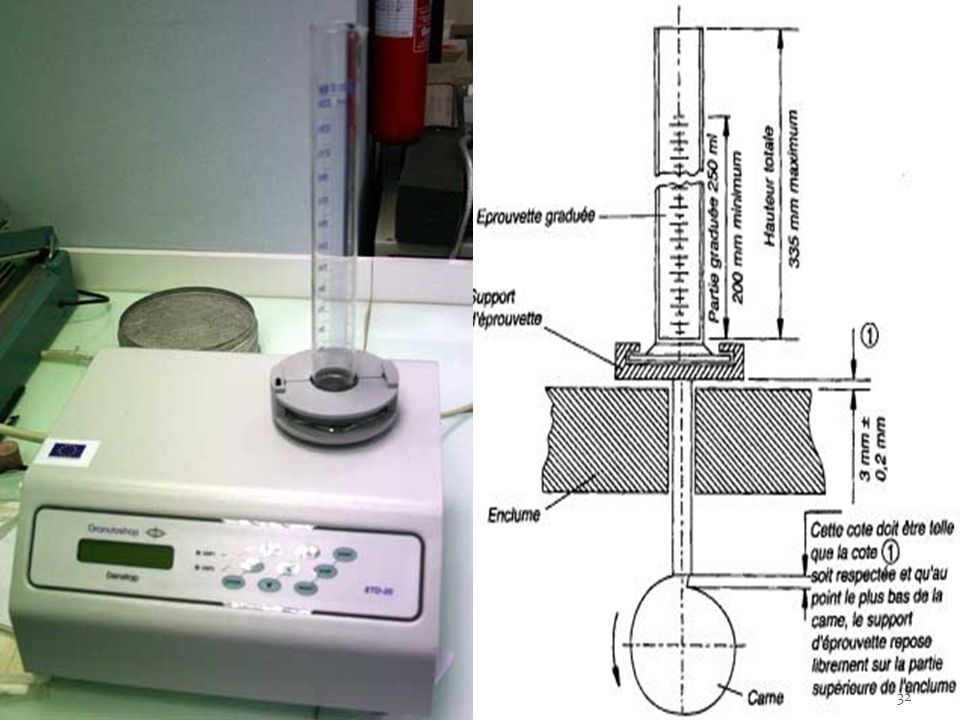
L’étude du tassement des poudres se fait sous très faibles contraintes de manière à analyser l’aptitude de la poudre à se réarranger. Elle consiste à déterminer dans des conditions bien définies, les volumes apparents occupés par une masse donnée avant et après tassement. La poudre doit se réarranger aisément puis se tasser le plus faiblement possible. -le test est réalisé à l’aide d’un voluménomètre de tassement équipé d’une éprouvette de 250 ml. 100 grammes de poudre versés dans l'éprouvette. Nb chutes: 0, 10, 20,, 100,, 500, 1000, L'essai est réalisé 3 fois. Les volumes sont notés V0 (volume vrac), V10, V20 …. V1250
, V10, V20 …. V1250.")
33
V500: volume après 500 tassements
Le tassement des poudres représente la capacité des particules à se réarranger spontanément ou sous l'effet de sollicitations mécaniques. Il permet de prévoir l'aptitude de la poudre à se réorganiser dans les matrices de compression. V0 : volume avant tassage V10: volume après 10 tassements. intérêt: régulariser le plan supérieur du lit, faciliter la lecture. V500: volume après 500 tassements V10 – V500 doit être < 20 ml : poudre comprimable directement.

34
Si V10 – V500 > 20 ml : mauvais écoulement et donc une phase de tassement importante pendant le cycle de compression. Revoir la formule, ou envisager une granulation.

35
C-3-c-La comprimabilité ou la cohésion
Lors des études de préformulation, l’une des questions importantes que se pose le formulateur est de savoir si un produit est susceptible de donner des comprimés par compression. C’est à ce niveau qu’intervient le test de comprimabilité et qui s’applique aussi bien à un principe actif seul ou mélangé à un excipient pour compression directe, qu’à un grain. Ce test permet également d’orienter le formulateur dans son choix d’excipient en lui évitant de faire d’inutiles essais avec certains excipients que l’on sait grâce aux tests incapables d’améliorer le mélange.

36
C-3-c-La comprimabilité ou la cohésion
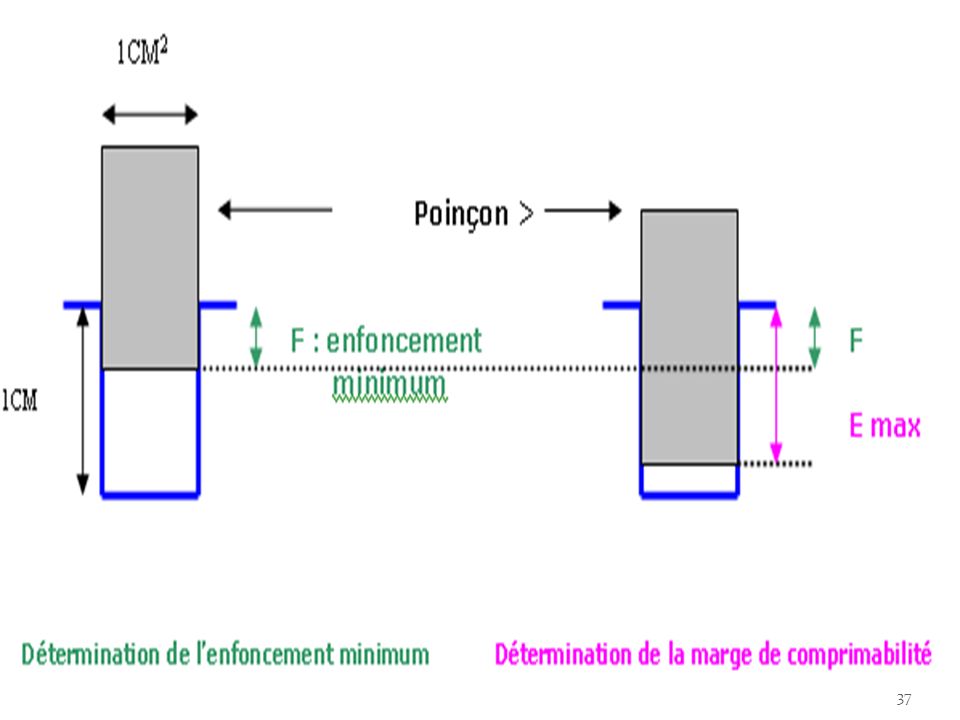
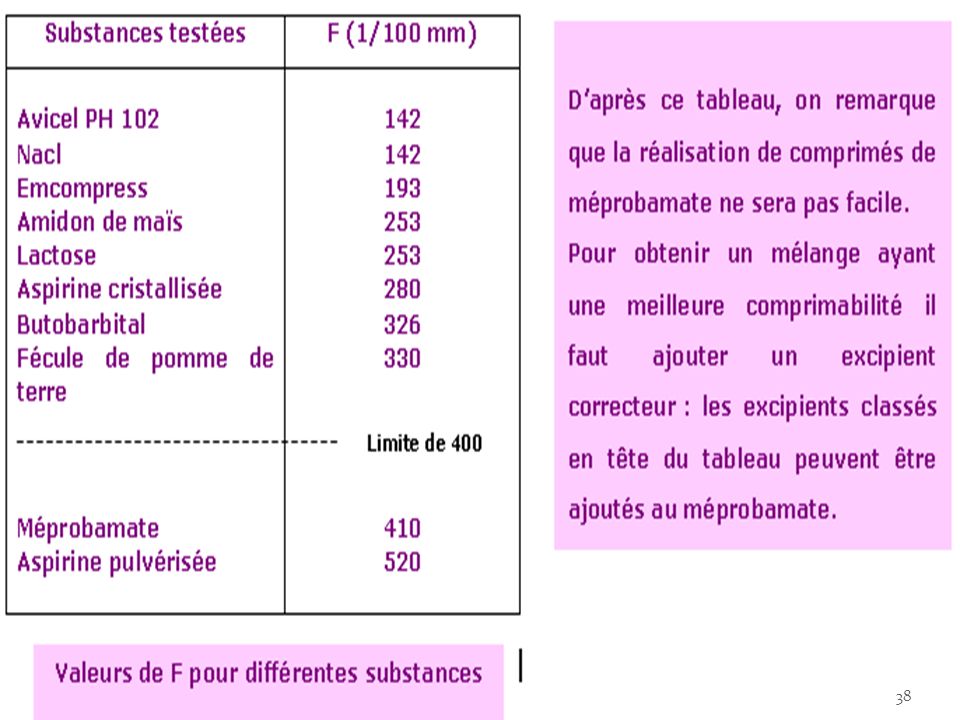
1- La comprimabilité d’un produit donné correspond à l’enfoncement minimum du poinçon supérieur permettant d’obtenir une colonne suffisamment dense pour présenter l’aspect extérieur d’un comprimé, mais de dureté nulle c'est-à-dire s’écrasant entre les doigts. pour une chambre de compression de 1 cm de profondeur et pour des poinçons de 1 cm2 de surface, si F exprimée en 1/100e de mm est supérieur à 400( soit 4mm), il est difficile d’obtenir des comprimés dans des conditions normales de fabrication, il faut donc ajouter un excipient correcteur à F<400.
, il est difficile d’obtenir des comprimés dans des conditions normales de fabrication, il faut donc ajouter un excipient correcteur à F<400.")
39
l’étude de la dureté des comprimés en fonction de l’enfoncement du poinçon supérieur dans la chambre de compression nous renseigne sur sa marge de comprimabilité. En effet, les limites extrêmes de possibilité de variation de l’enfoncement du poinçon supérieur se situent entre une valeur d’enfoncement minimal F donnant une dureté nulle et l’enfoncement maximal Emax au delà duquel la machine se bloque. L’optimisation consiste donc ici à choisir les mélanges qui donneront la marge de comprimabilité (F – E max) la plus grande.
 la plus grande.")
40
C-3-d-- La compressibilité
La compressibilité est l'aptitude de la poudre à acquérir de la cohésion lorsque la pression augmente. La caractérisation de la comprimabilité est basée d'une part sur l'analyse des cycles de compression et d'autre part sur la détermination des propriétés mécaniques des compacts élaborés. C’est l’étude des cycles de compression en utilisant : Des jauges de contrainte pour mesurer les contraintes Des capteurs à induction pour mesurer les déplacements. Le cycle de compression pour chaque comprimé fabriqué sera visualisé sur un écran cathodique.

41
d- Étude de stabilité Temps pendant lequel la substance conserve son intégrité sur les plans qualitatif et quantitatif . Les essais de stabilité sont menées dans des conditions déterminées de température, d’humidité et de lumière. - Le but en est d’établir les conditions de stockage, la date limite d’utilisation et de choisir un conditionnement approprié.

42
1- L’étude systématique de stabilité (Condition ICH: International Conference on Harmonisation ) Comporte : a- Essai de dégradation accélérée: Étude conçue pour accélérer la vitesse de dégradation chimique ou d’altération physique d’un produit médicamenteux ou d’une substance médicamenteuse active, qui fait appel à des conditions de stockage excessives. b-Essais de longue durée (en temp réel): Études de stabilité effectuées dans les conditions recommandées de stockage. c-Essais sous conditions intermédiaires: Études réalisées à 30°C/65 % HR dans lesquelles on augmente modérément la vitesse de dégradation chimique ou d’altération physique d’une substance médicamenteuse ou d’un produit médicamenteux destiné à être stocké pendant de longues périodes à 25°C.
: Études de stabilité effectuées dans les conditions recommandées de stockage. c-Essais sous conditions intermédiaires: Études réalisées à 30°C/65 % HR dans lesquelles on augmente modérément la vitesse de dégradation chimique ou d’altération physique d’une substance médicamenteuse ou d’un produit médicamenteux destiné à être stocké pendant de longues périodes à 25°C.")
43
Sub médt à être entreposées au réfrigérateur
Cas général Sub médt à être entreposées au réfrigérateur Etude Condition Durée Longue durée 25°C±2°C / 60%HR ± 5%HR Ou 30°C±2°C / 65%HR ± 5%HR 12 mois Conditions intermédiaires 30°C±2°C / 65%HR ± 5%HR 6 mois Dégradation accélérée 40°C±2°C / 75%HR ± 5%HR Etude Condition Durée Longue durée 5°C ± 3°C 12 mois Dégradation accélérée 25°C±2°C / 60%HR ± 5%HR 6 mois

44
3- Etude de la photostabilité
L’étude de la stabilité de la molécule à la lumière se fait sous forme de poudre et en solution. - La poudre est conservée dans un récipient constitué par une couche mince de verre, de 5cm de diamètre. Chaque récipient doit contenir 60 mg de la substance active étalé sur une épaisseur ne dépassant pas 3 mm. La poudre est exposée à la lumière artificielle (lampe au xénon) pendant 6 à 12heure. -En solution Une solution de la substance active de concentration 1mg/ml est répartie dans des ampoules en verre de 10 ml et elle est exposée à la lumière artificielle (lampe au xénon) pendant 6 à 12heures. La moitié des ampoules est couverte de papier aluminium et sert de témoin pour détecter d’éventuelle dégradation pendant l’exposition.
 pendant 6 à 12heure. -En solution. Une solution de la substance active de concentration 1mg/ml est répartie dans des ampoules en verre de 10 ml et elle est exposée à la lumière artificielle (lampe au xénon) pendant 6 à 12heures. La moitié des ampoules est couverte de papier aluminium et sert de témoin pour détecter d’éventuelle dégradation pendant l’exposition.")
45
Méthodologie étude stabilité nouveau PA
1- Prévision théorique de la réactivité de la molécule 2- Etude pratique :stabilité intrinsèque de la molécule Dégradation sévère :en solution aqueuses ou hydro-organiques : eau, milieu acide, alcalin et oxydant. Dégradation sévère sous forme de poudre : T° élevée, flacon ouvert et flacon fermé T° élevée, humidité relative élevée, flacon ouvert . Résultat : connaissance des pt fragiles molécule Produit de dégradation potentiel de la molécule

46
La stabilité chimique d’un PA au sein d’une forme galénique dépend de :
L’humidité : impliquée directement dans les R° d’hydrolyses ou favorise les R° d’oxydations et covalente. PH : influence sur l’intensité des R° d’hydrolyses et peut mettre le PA sous état d’ionisation ± réactif .

47
3- Compatibilité PA –excipients
Les interactions PA-excipient sont le résultat d’une : Incompatibilité chimique pd fabrication ou au cours stockage, il résulte :Dégradation intrinsèque du PA sans R° covalents avec les excipients ex : R°d’hydrolyse, oxydation………………

48
Dégradation consécutive à des R°covalentes : les amines Iaire et IIaire réagissent avec le lactose mais pas avec le mannitol Modification état physique du PA par : -Complexation -Précipitation -Modification de la forme cristalline Ces interactions ont un impact négatif sur stabilité chimique et la cinétique de dissolution et biodisponibilité .

49
Méthodologie choix excipient
Mélange PA +excipients Proportion habituelle d’une forme pharmaceutique 200mg de poudre au total +20%d’eau 50°C en flacon ou ampoule scellé analyse HPLC après 1 à 3 semaines.

50
Rôle de l’ajout de l’eau :
favorise mélange PA –excipient dégradation + importante que dans les conditions normales comparaison possible dans les m conditions entre divers excipients et choix de ceux qui dégradent le moins. permet de ne pas dépendre de l’hygroscopicité des excipients qui conditionne la qté d’eau localement disponible si on se contente d’exposer le mélange à une HR imposé.

51
L’étude se fait en plusieurs étapes :
PA diluant lubrifiant Lactose stéarate de magnésium Mannitol Acide stéarique Cellulose microcristalline PA + le meilleur couple diluant/lubrifiant +divers liant et désintégrant (*) Eventuellement le meilleur mélange obtenu en(*) colorant+agent d’enrobage+…..
 Eventuellement le meilleur mélange obtenu en(*) colorant+agent d’enrobage+…..")
52
Ex1 : PA = inhibiteur calcique BMS
Ester, amine tertiaire produit dégradation S/forme chlorhydrate Alcool L’alcool est le seul produit de dégradation en solution. L’alcool est le seul produit de dégradation observé lors des études de compatibilité

53
Résultat après 3 semaines à 50°C ,20%d’eau /Flacon scellé : Diluant
PA 200 mg PA/lactose mg PA/mannitol 25-175mg PA/phosphate dicalcique Teneur PA 96 ,4% 95,7% 95,8% 85,0% Teneur produit dégradât° 3,3% 4,1% 4,0% 16,7% PH - 6,2-6,5

54
Liants En solution, stabilité optimale du principe actif à ph =3.8
PA/lactose/ stéarate de magnésium PA/lactose /acide stéarique mg PA/mannitol / stéarate de magnésium /acide stéarique Teneur PA 64,3% 90,0% 65,4% 92,9% Teneur produit dégradât° 37.0% 9,7% 36,7% 6,9% PH 5 ,5 3 ,8 5,5 3,8

55
Ex2: PA = inhibiteur calcique BMS
Ester, amine tertiaire produit de dégradation Sous forme de chlorhydrate En mélange binaire(avec mannitol ou lactose ): peu de dégradation ,seul le produit I est formé (hydrolyse de fonction ester).
: peu de dégradation ,seul le produit I est formé (hydrolyse de fonction ester).")
56
En mélange tertiaire (PA+diluant +lubrifiant)
Mannitol ou lactose +Ac stéarique : peu de dégradat° ,seul le produit I est formé . Mannitol +stéarate de mg : dégradat° + importante (liée au PH ) ,seul le produit I est formé . - Lactose+ stéarate de Mg : dégradat°+importante, I et II sont observés cm produits de dégradat° Résultat de l’étude : -Seul les couples Mannitol +acide stéarique ou lactose+ Ac stéarique peuvent être utilisés . L’utilisation des mélange binaires ne suffit pas (interactions plus complexe)
 ,seul le produit I est formé . - Lactose+ stéarate de Mg : dégradat°+importante, I et II sont observés cm produits de dégradat° Résultat de l’étude : -Seul les couples Mannitol +acide stéarique ou lactose+ Ac stéarique peuvent être utilisés . L’utilisation des mélange binaires ne suffit pas (interactions plus complexe)")
57
II-2-2- La formulation II-2-2-1- Définition
II Objectif de la formulation a - Choix de la forme du PA b- Choix de la voie d’administration c- Choix de la forme galénique d- Choix de L’excipient d-1- Définition : d-2- Critères de choix d’un excipient - faisabilité pharmacotechnique e-Procédé de fabrication f- Le Conditionnement

58
II Définition La formulation est une activité industrielle, consistant à fabriquer des produits homogènes, stables et possédants des propriétés spécifiques, en mélangeant différentes matières premières, sous une forme pharmaceutique définie. PA + excipients (Matière première) + technologie = formulation II Objectif de la formulation - Générer des prototypes ce qui est lié : Aux spécifications du principe actif et celles de l’excipient ainsi qu’au procédé de fabrication :
 + technologie = formulation. II Objectif de la formulation - Générer des prototypes ce qui est lié : Aux spécifications du principe actif et celles de l’excipient ainsi qu’au procédé de fabrication :")
59
Au facteur économique (innovation) : médicament doit être rentable
Les caractéristiques physico-chimiques du PA, le moins d’excipients et les technologies les plus simples possibles Aux définitions des objectifs thérapeutiques : nature pathologie, activité du PA et facteurs inhérent au patients Au facteur économique (innovation) : médicament doit être rentable Aux méthodologies existantes. Aux systèmes experts d’aide à la formulation. - Optimisation de la formulation au fur et à mesure des résultats des différents essais.
 : médicament doit être rentable. Aux méthodologies existantes. Aux systèmes experts d’aide à la formulation. - Optimisation de la formulation au fur et à mesure des résultats des différents essais.")
60
a)- Choix de la forme du PA
Le PA peut exister sous plusieurs formes cristallines, ou sous forme de dérivés tels que sel, ester…. hydrates. Le choix est fonction du: Mode d’administration stabilité : la forme polymorphe est plus stable solubilité : PA doit être le plus soluble possible biodisponibilité du PA b)- Choix de la voie d’administration Ce sont les études de préformulation qui orientent le choix de la voie d’administration qui se fait en fonction de : La biodisponibilité du PA La vitesse d’action désiré, durée traitement, nombre de prise par jour… Type de malades : l’âge du patient, maladie hospitalière ou ambulatoire.
- Choix de la voie d’administration Ce sont les études de préformulation qui orientent le choix de la voie d’administration qui se fait en fonction de : La biodisponibilité du PA. La vitesse d’action désiré, durée traitement, nombre de prise par jour… Type de malades : l’âge du patient, maladie hospitalière ou ambulatoire.")
61
c)- Choix de la forme galénique
Le choix est lié à celui de la voie d’administration Le choix de la forme dépend : voie d’administration envisagée ; vitesse d’action (immédiate, prolongée) ; La solubilité du PA en milieu aqueux et non aqueux ; L’irritabilité de la substance ; La stabilité de la substance ; Condition de conservation de la forme galénique ; Demi –vie de la substance ; L’acceptance du patient
 ; La solubilité du PA en milieu aqueux et non aqueux ; L’irritabilité de la substance ; La stabilité de la substance ; Condition de conservation de la forme galénique ; Demi –vie de la substance ; L’acceptance du patient.")
62
d)-Choix de L’excipient
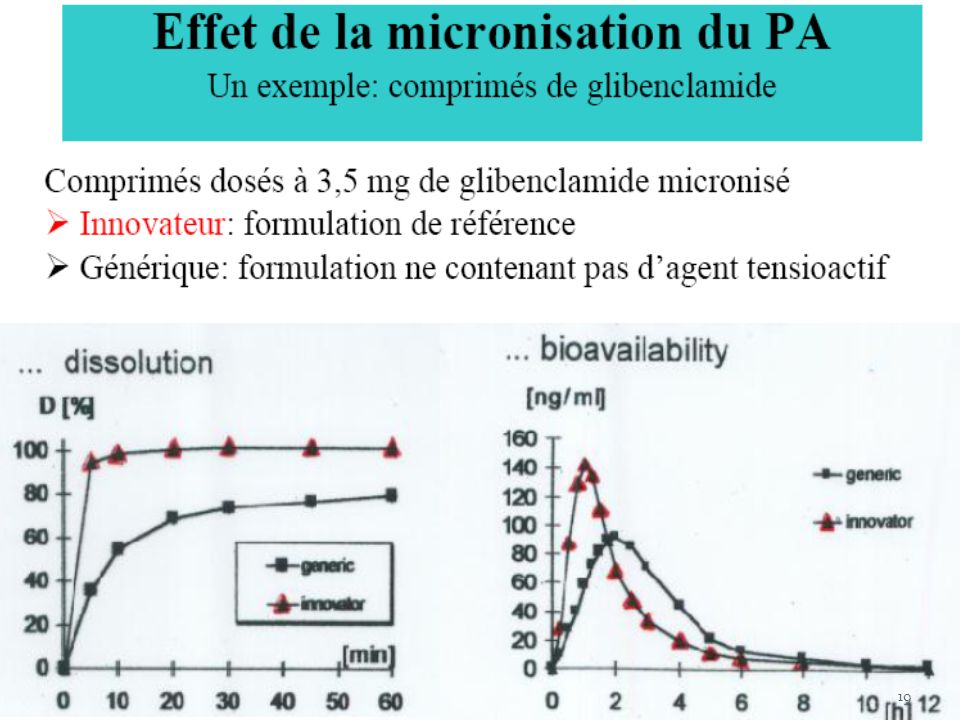
matière ou mélange de matières inactives sur la pathologie dépourvu donc de propriétés pharmacologiques, utilisé pour donner à 1 forme galénique 1 présentation convenable à son utilisation « poids, volume, goût, conservation, consistance». Ex : la vaseline donne la consistance à la pommade. Hormis le rôle de présentation, l’excipient permet de Maitriser : le temps de libération du PA(enrobage forme libération prolongée) ; Le lieu en ciblant la libération de la dose à un organe ou des cellules déterminées : les liposomes L’excipient à un rôle fondamental sur la biodisponibilité et la stabilité du principe actif ex : Phynotoine : remplacement sulfate de Ca par lactose ↑ vitesse de dissolution ↑ Complexation mort Gilbenclamide : ajout de tensioactifs pour ↑ vitesse de dissolution.
 ; Le lieu en ciblant la libération de la dose à un organe ou des cellules déterminées : les liposomes. L’excipient à un rôle fondamental sur la biodisponibilité et la stabilité du principe actif ex : Phynotoine : remplacement sulfate de Ca par lactose ↑ vitesse de dissolution ↑ Complexation mort. Gilbenclamide : ajout de tensioactifs pour ↑ vitesse de dissolution.")
63
Critères de choix d’un excipient
Vu le grand nombre d’excipient disponible, Le choix de l’excipient est assez difficile, basé sur les critères suivant : l’inertie vis-à-vis: des PA, matériaux de conditionnement et l’organisme (innocuité parfaite) La faisabilité pharmacotechnique La stabilité et compatibilité PA et excipients. L’inscription ou non a la pharmacopée L’influence sur les propriétés biopharmaceutique et technologique Considération économique (quantité et prix)
 La faisabilité pharmacotechnique. La stabilité et compatibilité PA et excipients. L’inscription ou non a la pharmacopée. L’influence sur les propriétés biopharmaceutique et technologique. Considération économique (quantité et prix)")
64
La faisabilité pharmacotechnique
La faisabilité est la qualité technologique de l’excipient ou rôle de l’excipient dans la formule. Dans la formulation des comprimés par exemple l’excipient doit favoriser l’aptitude à l’écoulement, à la cohésion, et à la dissolution. Ces propriétés orientent le galéniste dans le choix du procédé de fabrication : compression directe, granulation par voie humide……

65
Rôle de l’excipient Ex .excipient de première intention Diluant réduction % de PA ds mélange/grain et facilitent écoulement de la poudre lactose calcium hydrogène phosphate amidon de mais Liant permet fabrication du grain et ↑ dureté des Cp Cellulose microcristalline Amidon de mais prégélifie Polyvinyle polyvidone Hydroxypropyl cellulose Désagrégeant facilitent désagrégation du Cp et dissolution du PA Sodium carboxymethyl amidon ;Sodium carboxymethyl cellulose Lubrifiant d’écoulement favorisent écoulement du grain et éjection du Cp hors de la matrice Silice colloïdale Lubrifiant de compression réduction des frictions entre les particules pendant la compression donc : meilleure transmission de la force de compression Magnésium stéarate Palmito stéarate de glycérol

66
Ou lactose Fast-flow ou spray dry
Tableau : propriétés physico-technologique des différents types de lactoses. Type HHC grossier M M M M tamisé DCL Atomisé Ou lactose Fast-flow ou spray dry Principale application Tout domaine Granulation humide Compression directe Aptitude à l’écoulement Excellente Mauvaise Aptitude à la compression Moyenne Stabilité bonne Teneur en eau 5,2% 5%

67
Au cours de la compression les particules de matériaux pharmaceutiques se déforment principalement de manière élastique, plastique, fragmentaire : Les solides élastiques :Lors d'une déformation élastique, le solide retrouve sa forme initiale lorsque la force cesse d'être appliquée. Ce type de déformation réversible n'est donc pas favorable à l'obtention de comprimés, le matériau retrouvant son état initial en fin de compression.. Comportement à la compression du PA Comportement à la compression de l’excipient correcteur Exemple d’excipient à utilisé plastique Fragmentaire Lactose Calcium phosphate fragmentaire Plastique Cellulose microcristalline Élastique

68
Les solides plastiques sont caractérisées par la persistance de la déformation lorsque la force cesse d'être appliquée. Cette déformation s'accompagne de transfert de matière solide par écoulement interne. Ce phénomène irréversible est tout à fait favorable à l'obtention de comprimés de sorte que les matériaux plastiques sont recherchés comme excipients ou adjuvants de compression. Le chlorure de sodium, les celluloses microcristallines et les amidons sont les matériaux à déformation plastique les plus connus. Les solides fragmentaires ou fragiles sont des produits qui, sous l'application d'une contrainte, atteignent très rapidement leur seuil de rupture, avec une déformation plastique très faible. Le lactose, le saccharose et le phosphate de calcium appartiennent à cette catégorie.

69
e)- Procédé de fabrication e-1)-Définition :
Les procédés de fabrication ou de technologie pharmaceutique, sont des moyens techniques qui permettent de conférer aux substances et aux mélanges de substances les propriétés indispensables à la fabrication de forme pharmaceutiques. Chaque procédé se compose de plusieurs opérations pharmaceutiques : la granulation humide par exemple se décompose en : Mélange des poudres sèches Humidification et malaxage Granulation, séchage et calibration des granulés obtenus. Le choix d’un procédé spécifique garantit au produit sa qualité

70
e-2)- Choix du procédé de fabrication
Le choix du procédé de fabrication est fonction de : L’objectif à atteindre ou forme galénique désiré (CP, Sirop, Susp………). Le matériel industriel disponible, sa capacité et ses limites imposés par le constructeur : choix de machine robuste et économique. Prix de revient industriel. Les procédés de fabrication doivent être optimisés en fonction des matières premières, du matériel et des conditions opératoires : détermination des facteurs les plus influents sur la qualité du produit. La fabrication doit être robuste : résistante aux sensibilités dues aux variations minimes dans procédé de fabrication ex : variation dues au changement de fournisseur de matières premières, granulométrie, lot………..
. Le matériel industriel disponible, sa capacité et ses limites imposés par le constructeur : choix de machine robuste et économique. Prix de revient industriel. Les procédés de fabrication doivent être optimisés en fonction des matières premières, du matériel et des conditions opératoires : détermination des facteurs les plus influents sur la qualité du produit. La fabrication doit être robuste : résistante aux sensibilités dues aux variations minimes dans procédé de fabrication ex : variation dues au changement de fournisseur de matières premières, granulométrie, lot………..")
71
f)- Le Conditionnement
Le conditionnement est une opération complémentaire de la mise en forme pharmaceutique, qui assure la conservation du médicament pendant le temps prévu à son utilisation. Le conditionnement comporte : L’emballage primaire ou récipient en contact direct avec la substance médicamenteuse. L’emballage secondaire destiné à apporter une protection supplémentaire au produit.

72
Le choix du conditionnement primaire fait partie intégrante du médicament depuis le processus de développement à l’AMM. En dehors de ses qualité physique et mécanique (imperméabilité, étanchéité, résistance physique), un conditionnement primaire doit garantir l’innocuité et la qualité du médicament tous au long de son cycle de vie et faciliter son emploi. Le conditionnement pour solution injectable doit avoir en plus comme qualité la Transparence et la résistance hydrolytique et thermique . Le contrôle des interactions contenant –contenu est une exigence réglementaire aussi bien pour les nouveaux médicaments que pour le changement de conditionnement primaire pour toutes les formes pharmaceutiques déjà existantes. Et rentre dans le cadre des essais de stabilité.
, un conditionnement primaire doit garantir l’innocuité et la qualité du médicament tous au long de son cycle de vie et faciliter son emploi. Le conditionnement pour solution injectable doit avoir en plus comme qualité la Transparence et la résistance hydrolytique et thermique . Le contrôle des interactions contenant –contenu est une exigence réglementaire aussi bien pour les nouveaux médicaments que pour le changement de conditionnement primaire pour toutes les formes pharmaceutiques déjà existantes. Et rentre dans le cadre des essais de stabilité.")
73
Phénomènes d’interaction contenu-contenant
On distingue : L’adsorption : les molécules de médicaments sont adsorbées par le conditionnement primaire (ex matière plastique), il existe plusieurs facteurs pouvant exalter ce phénomène : - PH de la préparation pharmaceutique - le solvant qui peut avoir une action dissolvante facilitant la diffusion du soluté dans la matière plastique par exemple. La désorption : la préparation pharmaceutique au contact avec le contenant peut solubiliser certains additifs du matériau plastique qui migre (diffusion) dans le médicament et peuvent provoquer soit :
, il existe plusieurs facteurs pouvant exalter ce phénomène : - PH de la préparation pharmaceutique. - le solvant qui peut avoir une action dissolvante facilitant la diffusion du soluté dans la matière plastique par exemple. La désorption : la préparation pharmaceutique au contact avec le contenant peut solubiliser certains additifs du matériau plastique qui migre (diffusion) dans le médicament et peuvent provoquer soit :")
74
-Un problème de toxicité.
-Une altération de la formule pharmaceutique -Un problème de toxicité. Les matériaux autorisés dans la fabrication des récipients On distingue deux types de produits médicamenteux, plus ou moins susceptibles d’être modifiés par leur contenant : les produits injectables (préparations parentérales) et collyres : ils doivent être suivi de près par toutes les réglementations de la Pharmacopée. Ils sont très fragiles. les autres produits. Les médicaments et leur emballage doivent répondre à des tests bien précis. C’est pourquoi en général on retrouve quatre types de matériaux pour les emballages pharmaceutiques ,
 et collyres : ils doivent être suivi de près par toutes les réglementations de la Pharmacopée. Ils sont très fragiles. les autres produits. Les médicaments et leur emballage doivent répondre à des tests bien précis. C’est pourquoi en général on retrouve quatre types de matériaux pour les emballages pharmaceutiques ,")
75
répondant le mieux aux exigences alimentaires et à l’absence d’interaction avec le contenu. On trouve : les plastiques, le verre (ampoules, flacons, seringues..), l’aluminium (blisters, et tubes……………..)Et les élastomères (bouchage des flacons). 1- Récipients en matière plastique Les plastiques possèdent de multiples propriétés : transparence ou opacité, imperméabilité aux gaz, aux odeurs, possibilité de formes diverses, propreté et hygiène des manipulations, légèreté, prix de revient faible. Selon la Pharmacopée Européenne : " Les récipients en matières plastiques sont fabriqués à l’aide de matériaux constitués d’un ou plusieurs polymères, ainsi que d’additifs éventuels.

76
Ces matériaux ne comportent pas dans leur composition des substances qui pourraient être extraites par le contenu du récipient dans des proportions entraînant pour ce dernier une altération de son efficacité ou de sa stabilité ou une augmentation de sa toxicité. La nature des additifs et leur quantité sont fonction du type de polymère utilisé, du procédé de transformation du récipient et de l’usage du récipient. Ces additifs sont des antioxygènes, des stabilisants, des plastifiants, des lubrifiants, des colorants et des renforceurs mécaniques. Les agents antistatiques et de démoulage ne sont utilisés que pour des récipients pour des préparations à usage oral ou usage externe pour lesquels ils sont autorisés.

77
Des additifs acceptables sont indiqués dans la formulation type de chaque matériau décrit dans la pharmacopée. " 2- Récipients en verre Pour les emballages pharmaceutiques en verre, on emploie plusieurs types de qualité de verre selon le produit qui va être conditionné. Ces verres utilisés sont classés en fonction de leur résistance hydrolytique, c’est-à-dire la résistance du verre à la cession de substances minérales solubles dans l’eau, dans des conditions déterminées de contact entre la surface intérieure du récipient et de l’eau. Cette résistance est évaluée par le titrage par un acide de l’alcalinité de la solution (concentration en ions OH- qui corrodent le verre).
.")
78
Selon leur résistance hydrolytique et leur stabilité chimique, les récipients verre sont classés par la Pharmacopée comme suit : - verre de type I : verre dit " neutre dans la masse ", dont la résistance hydrolytique élevée est due à la composition chimique de la masse (verre borosilicaté) - verre de type II : verre de silicate de soude, dont la résistance hydrolytique élevée résulte d’un traitement de surface. - verre de type III : verre de silicate de soude de résistance hydrolytique moyenne (non résistant à l’eau : il n’est pas destiné à contenir des produits aqueux) - verre de type IV : verre de silicate de soude de résistance hydrolytique faible. Remarque : Seul le verre de type I est réutilisable
 - verre de type II : verre de silicate de soude, dont la résistance hydrolytique élevée résulte d’un traitement de surface. - verre de type III : verre de silicate de soude de résistance hydrolytique moyenne (non résistant à l’eau : il n’est pas destiné à contenir des produits aqueux) - verre de type IV : verre de silicate de soude de résistance hydrolytique faible. Remarque : Seul le verre de type I est réutilisable.")
79
II-2-3- Optimisation de la formulation
II Mise au point du prototype II Les essais cliniques. II Demande d’autorisation de mise sur le marché

80
II-2-3- Optimisation de la formulation
II Mise au point du prototype Proposer une formule Etude de stabilité Etude de la biodisponibilité Refaire les études avec une formule semi- quantitative Changement d’échelle Proposer une formule quantitative Prenant l’exemple des comprimés :

81
On propose plusieurs formules
On effectue plusieurs essais : L’aptitude à l’écoulement Volume de tassement Aptitude à la compression Formule 1 Formule 2 Formule 3 Formule 4 PA xg diluant ag bg cg dg liant wg yg zg délitant Lubrifiant d’écoulement Ag Bg Cg Dg Lubrifiant de compression ..g …g

82
En fonction des résultats obtenus on choisit la composition du mélange le plus performant.
On poursuit les essais après la compression : Tests galéniques : essai de désagrégation. Essai de résistance à la rupture. Après ces études le domaine réduit est sélectionné on passe alors aux : Domaine sélectionné à 1 Kg : refaire les tests précédents Domaine sélectionné à 7 Kg : refaire les essais Domaine sélectionné échelle pilote 20Kg : refaire les essais Obtention donc du prototype

83
II.2.3.3- Demande d’autorisation de mise sur le marché
La demande d’autorisation de mise sur le marché est adressée à l’autorité compétente, et comporte une description détaillée et précise du médicament. L’AMM reste valable 5 ans.

84
Constitution du dossier d’AMM :.
Partie I : partie pharmaceutique 1. Coordonnées du fabricant. 2. Dénomination Spéciale du médicament. 3. Composition qualitative et quantitative 4. Forme pharmaceutique, mode et voie d’administration. Posologie usuelle. 5. Description des conditions de conservation : Durée de conservation proposée, précautions particulières de conservation. 6. Description du procédé de fabrication et conditions de fabrication 7. Contrôles des matières premières et du produit fini. 8. Contrôles des articles de conditionnement. 9. Echantillon du modèle et des notices d’information.

85
Partie II : toxicologique
Études de toxicologie sur des modèles animaux ou biologiques afin de déterminer la sécurité d'emploi à des doses déterminées. Toxicité aiguë ; Toxicité chronique ; Potentiel mutagène ; Potentiel cancérogène ; Examen de la fonction de reproduction ; Toxicité embryofoetale et périnatale ; Tolérance locale. Partie III : pharmacologique et clinique : sécurité-efficacité

86
Pharmacologie : Pharmacodynamie : afin de repérer les modifications apportées au processus pathologique dont on recherche à limiter la progression ou les effets et d'en mesurer les bénéfices ; Interactions médicamenteuses ; Pharmacocinétique. Essais cliniques : Phases I, II et III. Toutes les phases des essais cliniques sont présentées : résumés tabulés des essais ; rapports exhaustifs des essais cliniques. Expériences après la mise sur le marché (phase IV) le cas échéant.
 le cas échéant.")
87
II.2.4- Production industrielle
La formulation aboutit à la confection du lot clinique ou prototype du lot industriel futur, qui doit être validé par les essais cliniques. Si les essais cliniques sont concluants, le prototype fait l’objet d’une demande d’autorisation de mise sur le marché (AMM). Une fois l’AMM accordé, le médicament sera produit à échelle industrielle et commercialisé.
. Une fois l’AMM accordé, le médicament sera produit à échelle industrielle et commercialisé.")
88
Les médicaments sont fabriqués par lots, à chacun des lots correspondent des fiches de fabrication sur lesquelles sont indiqués les différentes opérations effectuer : -Les pesés des matières premières -Les différentes étapes du processus de fabrication et de conditionnement avec indication des paramètres à respecter ex : vitesse du mélangeur et la durée du mélange. Pour pouvoir assurer la qualité du médicament et la reproductibilité du prototype ,il faut faire appel aux méthodes modernes de gestions de la qualité.

89
II.2.4- 1- Gestion de la qualité
L’entreprise doit disposer d’un système d’assurance qualité bien conçu, correctement mis en œuvre et efficacement contrôlé. L’assurance qualité se fait par l’application des bonnes pratiques de fabrication pharmaceutique qui permettent de maitriser La main- d’œuvre ; le matériel (locaux et équipement) ; le milieu ; les méthodes et les matières (matière première et article de conditionnement). « Règle des 5M »
 ; le milieu ; les méthodes et les matières (matière première et article de conditionnement). « Règle des 5M »")
90
Bonne pratique de la fabrication (BPF) : Les BPF pharmaceutiques sont constituées par l’ensemble des règles à mettre en œuvre pour la prévention des erreurs lors de la fabrication du médicament. Le personnel :Le fabricant doit disposer d’un personnel qualifié et en nombre suffisant pour mener à bien toutes les taches qui lui incombent. Les responsabilités individuelles doivent être clairement comprises par les intéressés. Tous les membres du personnel doivent être conscients des principes de Bonnes Pratiques de Fabrication qui les concernent; il convient d’assurer leur formation initiale et continue et notamment de donner les instructions d’hygiène en rapport avec l’activité exercée

91
Des programmes détaillés consacrés à l’hygiène doivent être établis et adaptés aux différents besoins de l’entreprise. Locaux et équipements : Les locaux doivent être situés, conçus, construits, adaptés et entretenus de façon à convenir au mieux aux opérations à effectuer. Leur conception doit tendre à minimiser les risques d’erreurs et à permettre un nettoyage et un entretien efficaces ,en vue d’éviter les contaminations croisées, le dépôt de saletés et, de façon générale, toute atteinte à la qualité des produits. Le matériel de fabrication et de contrôle doit être conçu et installé en fonction de sa destination, il doit être conçu de façon à permettre un nettoyage facile et minutieux. Il doit être nettoyé selon des procédures écrites détaillées et rangé dans un endroit propre et sec.

92
Documents : Les fabricants doivent utiliser un système de documentation couvrant les différentes opérations de fabrication effectuées. Ces documents retracent l'histoire de chaque lot produit. Des systèmes de traitement électroniques ou autres peuvent remplacer les documents écrits. Dans ce cas, le fabricant doit prouver que les données seront correctement conservées pendant la période envisagée. Production : La production doit être effectuée dans le respect des bonnes pratiques de fabrication et être conforme aux instructions et procédures préétablies.

93
Afin d'éviter notamment les contaminations croisées, des mesures à caractère technique ou organisationnel doivent être prises. Une unité de production est constituée par un ensemble de locaux délimités ,traversés par un flux de matières dont la qualité doit être parfaitement maitrisée .

94
Contrôle de la qualité Les fabricants doivent avoir à leur disposition un département de contrôle de la qualité. Ce département est placé sous l'autorité d'une personne indépendante des autres départements qui possède les qualifications requises. Il doit disposer d'un ou de plusieurs laboratoires de contrôle possédant le personnel et le matériel suffisants afin de procéder aux essais nécessaires sur les matières premières et articles de conditionnement, ainsi qu'aux contrôles sur les produits intermédiaires et finis. Le recours à des laboratoires extérieurs peut être autorisé. Lors de l'évaluation des produits finis avant leur libération pour la vente ou la distribution, le département de contrôle de la qualité doit notamment tenir compte des conditions de production, des résultats des contrôles en cours de fabrication, de l'examen des documents de fabrication et de la conformité des produits aux spécifications.

95
CONCLUSION Le galéniste est impliqué dans la conception du médt.IL doit prendre en considération toutes les caractéristiques du PA pour choisir qualitativement et quantitativement les excipients appropriés qui lui seront associés afin d'obtenir la forme pharmaceutique voulue, utilisable par le patient. Cette forme doit être stable sur le plan physicochimique et microbiologique, et permettre d’optimiser l'efficacité thérapeutique et la biodisponibilité du PA. Le formulateur doit éviter aussi les interactions contenant-contenu lors du conditionnement. Toute les étapes de conception doivent s’effectuer dans le respect des textes officiels de bonnes pratiques de fabrication afin d’assurer la qualité.

Présentations similaires






>")
>")
>")













