Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Méthodes quantiques

2
Utilisation des méthodes quantiques
Détermination de la structure électronique (orbitales, charges atomiques, potentiel électrostatique, aspects stéréoélectroniques) Détermination des énergies Détermination des géométries Recherche des états de transition Effets de solvant
 Détermination des énergies. Détermination des géométries. Recherche des états de transition. Effets de solvant.")
3
Ytot( x1,y1,z1, ……..xi,yi,zi,……… )
La fonction d’onde: fonction des coordonnées des noyaux et des électrons Ytot( x1,y1,z1, ……..xi,yi,zi,……… ) Solution de l’équation de Schrödinger HtotYtot = EYtot
 Solution de l’équation de Schrödinger HtotYtot = EYtot.")
4
Passage aux unités atomiques (u.a.):
1 u.a. = kcal mol-1

5
Ytot = Ynucl(coord. noyaux)Yel(coord. elec)
1ère approximation: Born-Oppenheimer: Les noyaux sont supposés fixes par rapport aux électrons Ytot = Ynucl(coord. noyaux)Yel(coord. elec) Potentiel nucléaire Constante pour un jeu de coordonnées nucléaires Equation de Schrödinger électronique HelYel = EelYel
Yel(coord. elec) Potentiel nucléaire. Constante pour un jeu de coordonnées nucléaires. Equation de Schrödinger électronique HelYel = EelYel.")
6
soit où et est une somme d’opérateurs monoélectroniques
est une somme de termes se rapportant à des paires d’électrons (termes biélectroniques) et
 et.")
7
Cette équation ne peut être résolue de façon exacte
que pour l’atome à un électron. Solution: n: nombre quantique principal l ≤ n-1: nombre quantique azimutal -l ≤ m ≤+l: nombre quantique magnétique l =0,1,2,3,4,……. sous-couches s,p,d,f,…..

8
l est associé au moment cinétique orbital m est associé à
Introduction du spin: L’interprétation de la structure fine du spectre de l’hydrogène nécessite d’introduire un moment cinétique intrinsèque à l’électron, le spin , dont la norme est définie par le nombre quantique ½: La composante Sz de ce moment est msħ ms peut prendre deux valeurs, ±1/2.

9
est appelée spinorbitale
La fonction d’onde de l’électron se met sous la forme d’un produit d’une fonction des coordonnées spatiales Fn,l,m(x,y,z) et d’une fonction des « coordonnées » de spin h(ms): est appelée spinorbitale
 et d’une fonction des « coordonnées » de spin h(ms): est appelée spinorbitale.")
10
Système polyélectronique:
On ne peut plus obtenir de solution analytique exacte de la fonction d’onde 2ème Approximation: Approximation orbitalaire Fonction d’onde totale exprimée comme un produit de fonctions monoélectroniques, c.a.d. dépendant chacune des coordonnées d’un seul électron. Ces fonctions monoélectroniques sont les spinorbitales j définies précédemment. Mais ce produit n’est pas un simple produit……….

11
Les spinorbitales sont les orbitales moléculaires du système.
(i) Les électrons sont indiscernables (ii) Les électrons sont des fermions: la fonction d’onde Ψ doit être antisymétrique par rapport à l’échange de deux électrons => Principe de Pauli (deux électrons ne peuvent pas avoir tous leurs nombres quantiques égaux) La fonction d’onde est prise comme un produit antisymmétrisé des spinorbitales, le déterminant de Slater. Les spinorbitales sont les orbitales moléculaires du système.
 Les électrons sont indiscernables. (ii) Les électrons sont des fermions: la fonction d’onde Ψ doit être antisymétrique par rapport à l’échange de deux électrons => Principe de Pauli (deux électrons ne peuvent pas avoir tous leurs nombres quantiques égaux) La fonction d’onde est prise comme un produit antisymmétrisé des spinorbitales, le déterminant de Slater. Les spinorbitales sont les orbitales moléculaires. du système.")
12
orbitale électron Energie associée au déterminant de Slater:

13
Jij = intégrale biélectronique coulombienne
l’intégration porte sur un électron quelconque, ici l’électron 1 l’intégration porte sur deux électrons quelconques, ici les électrons 1 et 2 Jij = intégrale biélectronique coulombienne Kij = intégrale biélectronique d’échange

14
Ce qui amène aux équations de Hartree-Fock
On applique alors une méthode de résolution variationnelle: le meilleur choix de y est obtenu en minimisant l’énergie E par rapport à l’ensemble des paramètres définissant les orbitales moléculaires, avec la contrainte que les spinorbitales sont orthonormales, c.a.d. Ce qui amène aux équations de Hartree-Fock (équation aux valeurs propres de l’opérateur de Fock F):
:")
15
Opérateur de Coulomb Opérateur d’échange

16
D’où la nécessité d’une procédure itérative
Donc F s’exprime en fonction des OM qu’on recherche D’où la nécessité d’une procédure itérative Dans la pratique, on part d’un ensemble d’orbitales moléculaires ji d’essai, on construit le déterminant de Slater, on calcule l’énergie correspondante. Cela implique le calcul des intégrales monoélectroniques hii et des intégrales biélectroniques Jij et Kij. On diagonalise F, ce qui fournit un nouvel ensemble d’orbitales ji qui sont fonctions propres de F, et on répète le processus jusqu’à convergence.

17
Résolution des équations de Hartree-Fock:
- soit numérique (possible uniquement avec les atomes) - soit analytique Résolution analytique en utilisant une 3ème Approximation: Approximation LCAO-MO: Chaque orbitale moléculaire s’exprime comme une combinaison linéaire de fonctions de base cp Ce sont les coefficients cpi qui sont les paramètres variationnels du calcul HF
 - soit analytique. Résolution analytique en utilisant une. 3ème Approximation: Approximation LCAO-MO: Chaque orbitale moléculaire s’exprime comme une combinaison linéaire de fonctions de base cp. Ce sont les coefficients cpi qui sont les paramètres variationnels du calcul HF.")
18
1ère approximation: Born-Oppenheimer
Equation de Schrödinger électronique HelYel = EelYel 2ème Approximation: Approximation orbitalaire = déterminant de Slater Méthode de résolution variationnelle Equations de Hartree-Fock 3ème Approximation: Approximation LCAO-MO

19
Approximation LCAO-MO:
Chaque orbitale moléculaire s’exprime comme une combinaison linéaire de fonctions de base cp Ce sont les coefficients cpi qui sont les paramètres variationnels du calcul HF Dans les programmes standards les fonctions de base cp sont des fonctions centrées sur les atomes.

20
Choix des fonctions de bases
3 considérations à prendre en compte: (i) La nature des fonctions doit avoir un sens d’un point de vue physique; (ii) Il faut choisir une forme de fonctions qui permet un calcul efficace des intégrales qui apparaissent dans les équations HF; (iii) Il faut garder un nombre de fonctions minimum (à cause de la progression de la longueur du calcul en N4);
 La nature des fonctions doit avoir un sens d’un point de vue physique; (ii) Il faut choisir une forme de fonctions qui permet un calcul efficace des intégrales qui apparaissent dans les équations HF; (iii) Il faut garder un nombre de fonctions minimum (à cause de la progression de la longueur du calcul en N4);")
21
(i) Choix naturel: fonctions de Slater du type
(ii) Cependant, les fonctions de Slater ne permettent pas un calcul aisé des intégrales, d’où l’idée de les remplacer par des fonctions gaussiennes de type en coordonnées polaires ou en coordonnées cartésiennes
 Cependant, les fonctions de Slater ne permettent pas un calcul aisé des intégrales, d’où l’idée de les remplacer par des fonctions gaussiennes de type en coordonnées. polaires ou en coordonnées cartésiennes.")
22
(iii) Le choix de fonctions gaussiennes conduirait, pour avoir des résultats pertinents, à un nombre total de fonctions très élevé, et par conséquent à un nombre d’intégrales également très élevé, d’où un coût de calcul rédhibitoire. On a donc été conduit à regrouper ces fonctions de base dites « primitives » en groupes de fonctions dites « contractées », chaque fonction contractée étant une combinaison linéaire de fonctions primitives. Les cp intervenant dans les orbitales moléculaires sont donc les fonctions contractées

23
• fonction de base « contractée »:
On aboutit donc à la hiérarchisation suivante: • gaussienne « primitive »: où lx + ly +lz détermine le type de fonction: lx + ly +lz = 0: fonction de type s lx + ly +lz = 1: fonction de type p lx + ly +lz = 2: fonction de type d etc… • fonction de base « contractée »: • orbitale moléculaire construite sur la base des fonctions contractées

24
La flexibilité de la base peut être augmentée par des fonctions de polarisation et des fonctions diffuses. Fonctions de polarisation: fonctions ayant un nombre quantique azimutal supérieur à celui des orbitales de valence, c.a.d. des orbitales p pour H, d pour C, N, O, f pour les métaux de transition. Fonctions diffuses: fonctions ayant un exposant très petit. Servent à décrire les régions loin du noyau, nécessaires pour décrire des anions.

25
1ère approximation: Born-Oppenheimer
Equation de Schrödinger électronique HelYel = EelYel 2ème Approximation: Approximation orbitalaire = déterminant de Slater Méthode de résolution variationnelle Equations de Hartree-Fock 3ème Approximation: Approximation LCAO-MO

26
E(n), non E(n-1) – E(n) ≤ seuil oui Choix de la base
Choix de la géométrie Choix des OM d’essai → Résolution des équations de Hartree-Fock E(n), non suffisamment semblable à E(n-1) – E(n) ≤ seuil oui Fin du calcul, convergence
, non. suffisamment semblable à. E(n-1) – E(n) ≤ seuil. oui. Fin du calcul, convergence.")
27
L’analyse de population
selon Mulliken

28
La densité électronique (c. a. d
La densité électronique (c.a.d. la probabilité de trouver un électron) d’une spinorbitale moléculaire à une position est donnée par le carré de cette spinorbitale or
 d’une spinorbitale moléculaire à une position. est donnée par le carré de cette spinorbitale. or.")
29
Si on somme sur toutes les spinorbitales moléculaires occupées et on intègre sur tout l’espace, on obtient le nombre total N d’électrons On généralise en introduisant un nombre d’occupation ni pour chaque spinorbitale moléculaire et en sommant sur toutes les spinorbitales

30
La matrice D dont les éléments sont Dpq est appelée matrice densité
Dans l’analyse de population selon Mulliken on utilise la matrice produit (D.S) pour déterminer les contributions atomiques des électrons. On a des contributions spécifiques à chaque fonction de base atomique, et des contributions partagées entre deux fonctions de base.
 pour déterminer les contributions atomiques des électrons. On a des contributions spécifiques à chaque fonction de base atomique, et des contributions partagées entre deux fonctions de base.")
31
(i) Elément diagonal: (ii) Elément non diagonal: C’est la moitié du nombre d’électrons partagés entre cp et cq
 Elément diagonal: (ii) Elément non diagonal: C’est la moitié du nombre d’électrons partagés entre cp et cq.")
32
On somme ensuite pour chaque atome l’ensemble des contributions à cet atome. Cela exige d’imposer un schéma de partition des contributions extradiagonales impliquant des fonctions de base localisées sur des atomes différents. Mulliken a proposé une équipartition entre les deux atomes impliqués. D’où: charge électronique de l’atome A: charge de l’atome:

33
Population de recouvrement entre deux atomes A et B:
L’analyse de population selon Mulliken a des limites dues à la partition arbitraire entre les contributions à deux atomes différents

34
Inconvénients de l’analyse de population suivant Mulliken
Éléments diagonaux peuvent être supérieurs à 2! Éléments non diagonaux peuvent devenir <0 ! Pas de justification pour le partage égal des contributions non diagonales entre deux atomes. Les plus électronégatifs devraient recevoir une contribution plus importante. Lorsqu’on a de très petits exposants sur des fonctions centrées sur un atome, ces fonctions vont décrire la fonction d’onde loin de cet atome. La partition de Mulliken ré-attribue la densité correspondante à l’atome, ce qui peut conduire à des effets bizarres. N’utiliser cette analyse de population que pour des comparaisons.

35
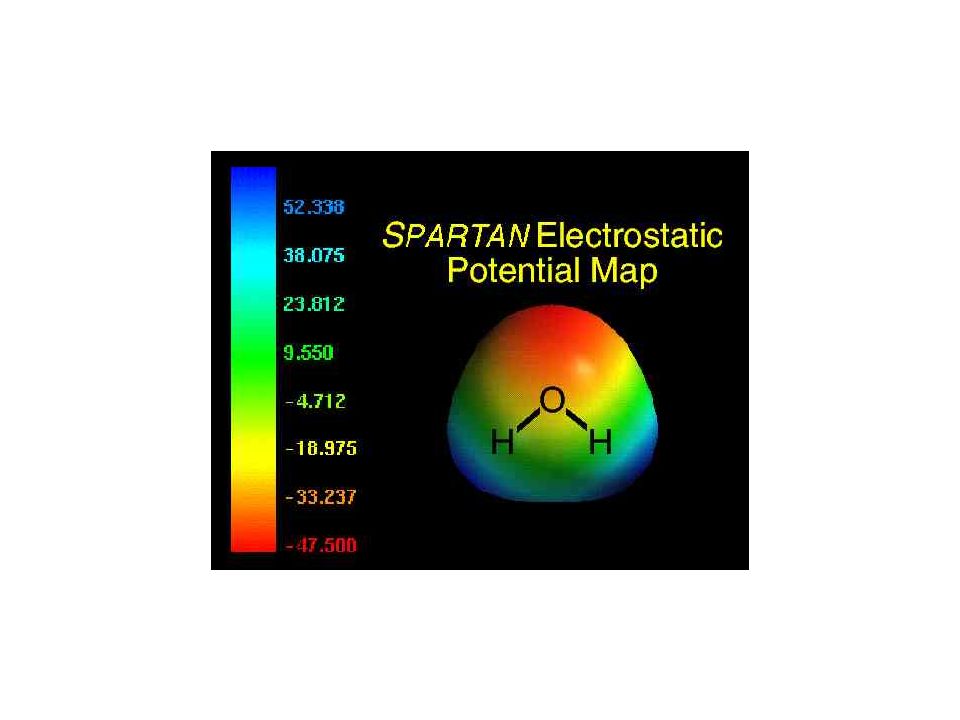
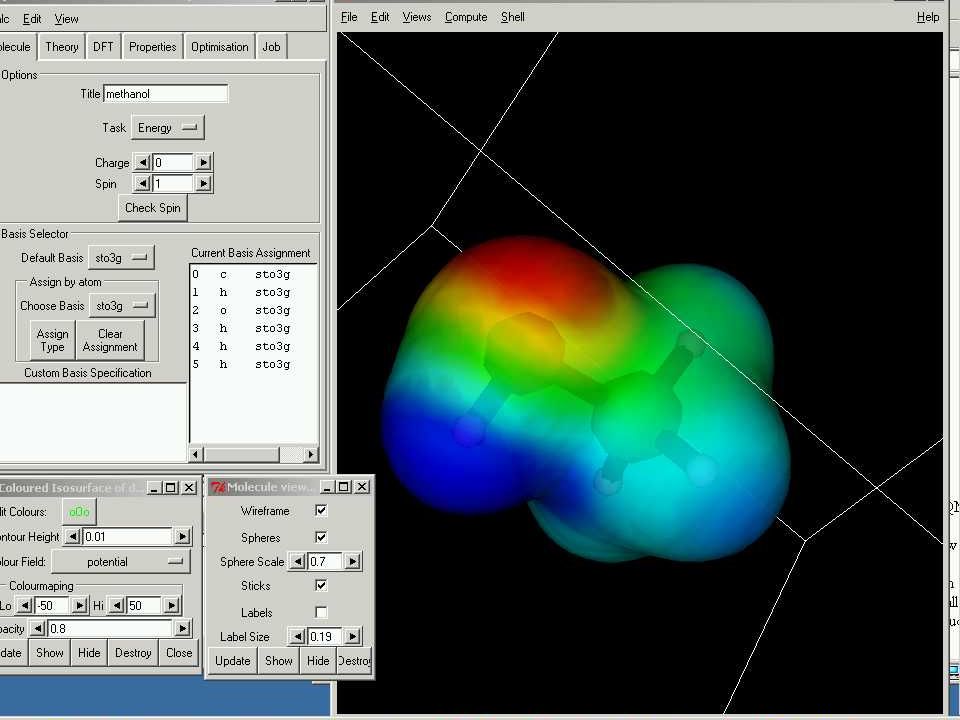
Le potentiel électrostatique

36
+ • q q0 Energie potentielle électrostatique à de l’origine et à de la charge q: En u.a. : Peut s’écrire: Où est le potentiel créé en par la charge q située en q1 • q2 +

37
Cas d’une molécule: Cas d’une distribution continue de charges:
- contribution des noyaux: - contribution de la densité électronique:

40
Analyse de population basée sur le potentiel électrostatique
On calcule le potentiel électrostatique de la molécule à partir de sa fonction d’onde, et on détermine les charges par ajustement entre ce potentiel électrostatique et un potentiel électrostatique obtenu à partir des charges, avec la contrainte que la somme des charges est égale à la charge totale de la molécule. Des contraintes supplémentaires peuvent être incluses, par exemple la reproduction du moment dipolaire.

41
Approximation de Born-Oppenheimer
Méthodes de type fonction d’onde E=F[Y(r)] Equation de Schrödinger électronique HelYel = EelYel Approximation orbitalaire: = déterminant de Slater Méthode de résolution variationnelle Equations de Hartree-Fock Approximation LCAO-MO Addition de déterminants supplémentaires Approximations supplémentaires Méthodes semi-empiriques (EHT, MNDO, AM1, PM3) Méthodes CI, MC-SCF, MPn,CC Vers la solution exacte
] Equation de Schrödinger électronique HelYel = EelYel. Approximation orbitalaire: = déterminant de Slater. Méthode de résolution variationnelle. Equations de Hartree-Fock. Approximation LCAO-MO. Addition de déterminants. supplémentaires. Approximations supplémentaires. Méthodes semi-empiriques. (EHT, MNDO, AM1, PM3) Méthodes CI, MC-SCF, MPn,CC. Vers la solution exacte.")
42
Au-delà de Hartree Fock

43
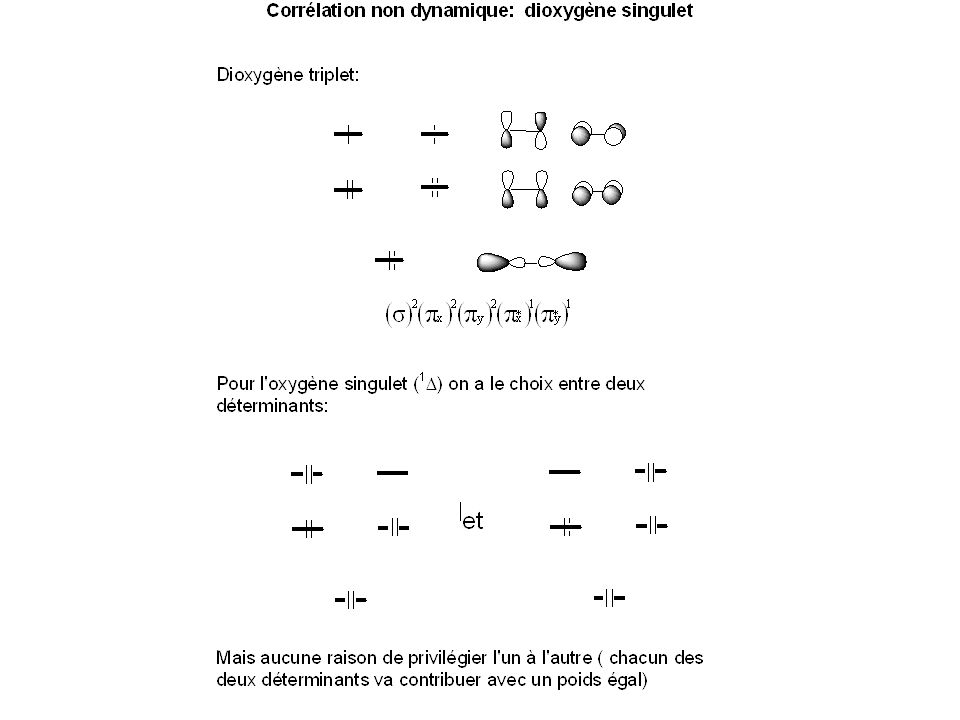
La corrélation électronique
Dans la méthode HF, où la fonction d’onde est un déterminant de Slater, chaque électron voit tous les autres comme un champ approché. La méthode HF ne rend pas compte du mouvement instantané des électrons => HF ne rend pas compte de la corrélation électronique, notamment pour les électrons de spins opposés Question: comment modifier la fonction d’onde HF pour obtenir une énergie plus basse? Les méthodes utilisées reposent sur le principe de la superposition de configurations

44
a0 >>> a1, a2, a3,….: corrélation dynamique
a0 ≈ a1, a2, a3,……: corrélation non dynamique

46
Les méthodes de résolution
• méthodes CI • méthodes MC-SCF • méthodes perturbationnelles: MPn • méthodes « Coupled Cluster »

51
Les méthodes CI et MC-SCF sont des méthodes variationnelles: on est sûr d’obtenir une limite supérieure de l’énergie exacte. Par contre avec les méthodes MPn, rien ne garantit que l’on obtienne une borne supérieure de l’énergie de corrélation. De plus la convergence, en termes d’ordre de perturbation, est aléatoire. Néanmoins la méthode MP2 et plus généralement les méthodes MPn restent une méthode de choix, notamment lorsqu’on qu’on doit traiter des interactions faibles. Pour la description de systèmes standard elle a cependant été « détrônée » par les méthodes DFT

52
Méthodes « Coupled Cluster »
agissant sur la fonction d’onde HF génère tous les déterminants ayant i excitations et

53
On peut en théorie inclure toutes les excitations d’un type donné (i=1,2,3,…) jusqu’à un ordre infini du développement de Taylor. En fait, on doit se limiter et la troncature s’effectue au niveau de l’opérateur T. Exemples: CCSD: CCSDT: L’effet de T3 peut être évalué par une méthode de perturbation: CCSD(T)
")
54
Approximation de Born-Oppenheimer
Méthodes de type fonction d’onde E=F[Y(r)] Equation de Schrödinger électronique HelYel = EelYel Approximation orbitalaire: = déterminant de Slater Méthode de résolution variationnelle Equations de Hartree-Fock Approximation LCAO-MO Addition de déterminants supplémentaires Approximations supplémentaires Méthodes semi-empiriques (EHT, MNDO, AM1, PM3) Méthodes CI, MC-SCF, MPn,CC Vers la solution exacte
] Equation de Schrödinger électronique HelYel = EelYel. Approximation orbitalaire: = déterminant de Slater. Méthode de résolution variationnelle. Equations de Hartree-Fock. Approximation LCAO-MO. Addition de déterminants. supplémentaires. Approximations supplémentaires. Méthodes semi-empiriques. (EHT, MNDO, AM1, PM3) Méthodes CI, MC-SCF, MPn,CC. Vers la solution exacte.")
55
Méthodes de la fonctionnelle de la densité
E=F[r(r)]
]")
56
Mais……………deux problèmes
E=F[r(r)] Idée sous-jacente: partir d’une observable physique pour déterminer l’énergie Théorème d’existence de Hohenberg et Kohn (1964): l’énergie d’un système, dans son état fondamental est complètement déterminée par la densité électronique r(r) Cette densité électronique obéit aussi au principe variationnel Mais……………deux problèmes Comment choisir de façon rationnelle des densités toujours meilleures? (ii) On ne connaît pas la forme analytique reliant l’énergie à la densité.
] Idée sous-jacente: partir d’une observable physique pour déterminer l’énergie. Théorème d’existence de Hohenberg et Kohn (1964): l’énergie d’un système, dans son état fondamental est complètement déterminée par la densité électronique r(r) Cette densité électronique obéit aussi au principe variationnel. Mais……………deux problèmes. Comment choisir de façon rationnelle des densités toujours meilleures (ii) On ne connaît pas la forme analytique reliant l’énergie à la densité.")
57
Décomposition de l’énergie:
E[r] = T[r] + Ene[r] +Eee[r] T: énergie cinétique des électrons Ene: énergie d’attraction électrons-noyaux Eee: énergie de répulsion électrons-électrons.

59
E[r] = Tni[r] + Ene[r] + J[r] + Exc[r]
DJ[r], K[r] et DT[r] sont inclus dans Exc[r] L’artifice du système fictif d’électrons non-interagissants, mais ayant la même densité que le système réel permet de réintroduire un déterminant de Slater composé de spinorbitales ji appelées orbitales de Kohn-Sham. Si on sait définir Exc, on a simplement un problème variationnel à résoudre: comme dans la méthode HF on minimise E via des équations de type HF. Comme dans la méthode HF on développe les orbitales de Kohn-Sham sur une base de fonctions.
![E[r] = Tni[r] + Ene[r] + J[r] + Exc[r]](http://slideplayer.fr/slide/1141758/3/images/59/E%5Br%5D+%3D+Tni%5Br%5D+%2B+Ene%5Br%5D+%2B+J%5Br%5D+%2B+Exc%5Br%5D.jpg "DJ[r], K[r] et DT[r] sont inclus dans Exc[r] L’artifice du système fictif d’électrons non-interagissants, mais ayant la même densité que le système réel permet de réintroduire un déterminant de Slater composé de spinorbitales ji appelées orbitales de Kohn-Sham. Si on sait définir Exc, on a simplement un problème variationnel à résoudre: comme dans la méthode HF on minimise E via des équations de type HF. Comme dans la méthode HF on développe les orbitales de Kohn-Sham sur une base de fonctions.")
66
Les méthodes semi-empiriques

67
Approximations communes
Seuls les électrons de valence sont considérés Base minimale, décrite par des fonctions de Slater Approximation ZDO (Zero Differential Overlap): (On néglige tous les produits de fonctions de base dépendant de la même coordonnée électronique si ces fonctions sont localisées sur des atomes différents A et B)
: (On néglige tous les produits de fonctions de base dépendant de la même coordonnée électronique si ces fonctions sont localisées sur des atomes différents A et B)")
68
Du fait de cette approximation, certaines intégrales mono et bi électroniques, en particulier les intégrales biélectroniques à 3 et 4 centres sont nulles Nécessité d’introduire des paramètres pour «compenser» ces approximations et obtenir des résultats «raisonables» Les acronymes correspondent à des méthodes différant par la façon dont la paramétrisation est effectuée

69
MNDO: Répulsion entre atomes distants de 2 à 3 Å en général trop grande: Molécules stériquement encombrées trop instables; Energies d’activation pour rupture ou formation de liaisons trop élevées Molécules hypervalentes trop instables Structures non classiques (e.g. cation alkyl) trop instables par rapport aux structures classiques Interactions faibles pas fiables; on ne peut pas prédire les liaisons hydrogène Et bien d’autres défauts…..
 trop instables par rapport aux structures classiques. Interactions faibles pas fiables; on ne peut pas prédire les liaisons hydrogène. Et bien d’autres défauts…..")
70
AM1: Bien meilleure: restent des défauts, par exemple:
Energies d’activation pour rupture ou formation de liaisons bien améliorées, restent cependant généralement trop élevées, et les géométries sont souvent trop «serrées» Molécules hypervalentes améliorées, mais il reste des erreurs plus grandes que sur les autres types de systèmes Groupes alkyles en général trop stables Force des liaisons hydrogènes prédite à peu près correctement, mais géométrie souvent fausse restent des défauts, par exemple: Liaisons peroxide trop courtes, géométries des composés avec des phosphores, problème conformationnel pour l’éthanol

71
PM3: S’appuie sur un très grand nombre de données expérimentales
Donne en général de meilleures énergies et de meilleures géométries, mais restent des problèmes d’analyse conformationnelle (e. g. éthanol, hydrazine) Liaisons hydrogènes encore trop courtes d’environ 0.1Å Liaisons de van der Waals mal décrites Liaisons Si-X (X=Cl, Br, I) sous-estimées Difficultés patentes à décrire l’azote, en général obtenu trop pyramidalisé Rotation peptidique (rotation autour de la liaison C-N dans les amides) sous-estimée ( parfois de 15 kcal/mol)
 Liaisons hydrogènes encore trop courtes d’environ 0.1Å. Liaisons de van der Waals mal décrites. Liaisons Si-X (X=Cl, Br, I) sous-estimées. Difficultés patentes à décrire l’azote, en général obtenu trop pyramidalisé. Rotation peptidique (rotation autour de la liaison C-N dans les amides) sous-estimée ( parfois de 15 kcal/mol)")
72
Prudence quand on utilise ces méthodes, les erreurs sur les énergies étant en général très aléatoires Méthodes semi-empiriques ont les mêmes avantages et inconvénients que les méthodes à champ de force: marchent relativement bien pour des structures proches des structures connues, mais elles sont souvent incapables de prédire des types de structures inconnues, car elles n’ont pas été paramétrisées pour cela. Cependant on peut les utiliser (mais sans garantie!) pour des études de réactivité Aucune possibilité d’amélioration systématique
 pour des études de réactivité. Aucune possibilité d’amélioration systématique.")
73
Optimisation des géométries

74
1-dimension E = constante de force x
Au voisinage du point d’équilibre: 1-dimension E = constante de force x • dérivée première de l’énergie est nulle: point stationnaire • énergie minimum => k>0

75
1-dimension aux points stationnaires k<0 maximum de l’énergie
(fréquence imaginaire, c.a.d. nombre complexe imaginaire) E k>0 minimum de l’énergie x = coordonnée de réaction A + B-C [A…..B…..C]‡ A-B + C
 E. k>0 minimum de l’énergie. x = coordonnée de réaction. A + B-C. [A…..B…..C]‡ A-B + C.")
76
2-dimensions équivalent à:

77
On peut développer l’énergie en une série de Taylor:
N-dimensions On peut développer l’énergie en une série de Taylor: : déplacement cartésien à partir du point de référence ; le gradient de l’énergie est nul Pour un point stationnaire = Matrice des constantes de forces = matrice hessienne

78
On peut réécrire E sous la forme:
soit On a alors soit La matrice C est la matrice des vecteurs propres de H ayant les l comme valeurs propres : modes normaux de vibration

79
Qi li > 0 pour tous les i => minimum d’énergie
N-dimensions i = 1, 2, ………3N-6 Points stationnaires: i = 1, 2, ………3N-6, c.a.d. grad E = 0 diagonalisation Qi li > 0 pour tous les i => minimum d’énergie lq < 0 , li > 0 pour tous les i différents de q => état de transition Qk : chemin de réaction à l’état de transition

80
Thermochimie

82
Les effets de solvant

83
Plusieurs modèles peuvent être utilisés:
(i) Modèle discret Molécule de solvant Molécule de soluté Inconvénients: • approche rapidement prohibitive en termes de durée de calcul; • incertitude quant à la pertinence de la géométrie optimisée
 Modèle discret. Molécule de solvant. Molécule de soluté. Inconvénients: • approche rapidement prohibitive en termes de durée de calcul; • incertitude quant à la pertinence de la géométrie optimisée.")
84
(ii) Modèle du continuum
Le soluté est considéré comme une distribution de charges électriques placée dans une cavité immergée dans un milieu continu de constante diélectrique e cavité soluté Cette distribution polarise le milieu, => induit un champ électrique R dit de réaction, qui en retour interagit avec la distribution de charges du soluté, ce qui conduit à une stabilisation. R dépend de e, de la forme et de la dimension cavité, et de la distribution de charges (ce qui conduit à un processus itératif sur cette distribution de charges)
")
85
Inconvénients des méthodes du continuum:
La distribution de charge du soluté peut être décrite par un dipôle moléculaire, ou par une distribution multipolaire. Dans le cas du modèle PCM (Polarized Continuum Model), la polarisation du diélectrique par le soluté est représentée par la création d’une distribution de charges apparentes sur la surface de la cavité. Inconvénients des méthodes du continuum: • l’énergie de solvatation dépend des paramètres de la cavité (taille, forme) • problèmes possibles d’adéquation de la géométrie du soluté à la géométrie de la cavité => utilisation d’une cavité obtenue à partir des sphères de Van der Waals des atomes du soluté se généralise
, la polarisation du diélectrique par le soluté est représentée par la création d’une distribution de charges apparentes sur la surface de la cavité. Inconvénients des méthodes du continuum: • l’énergie de solvatation dépend des paramètres de la cavité (taille, forme) • problèmes possibles d’adéquation de la géométrie du soluté à la géométrie de la cavité => utilisation d’une cavité obtenue à partir des sphères de Van der Waals des atomes du soluté se généralise.")
86
(iii) Modèle du semi-continuum (ou mixte)
Molécule de la première couche de solvatation continuum cavité soluté Inconvénients: taille du calcul, optimisation de géométrie peut être délicate

Présentations similaires












 Méthodes d’approximation>")


 Formalisme Quantique>")


 : I. Propriétés physiques des roches : densités, aimantations induites et.>")
 La modélisation moléculaire : optimisation.>")
 : I. Propriétés physiques des roches : densités, aimantations induites et.>")










