Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
DR. SIFI DR. REDOUANE-SALAH Hind
Unité motrice

2
Définition L'unité motrice : du motoneurone à la fibre musculaire Une unité motrice est formée par : le corps d’une cellule nerveuse motrice (neurone moteur ou motoneurone) situé dans la moelle épinière, son prolongement (axone) qui chemine dans le nerf périphérique et l’ensemble des fibres musculaires que le motoneurone innerve.
 situé dans la moelle épinière, son prolongement (axone) qui chemine dans le nerf périphérique. et l’ensemble des fibres musculaires que le motoneurone innerve.")
3
L’ensemble formé par un motoneurone α et la fibre musculaire qu’il innerve constitue ce que l’on appelle une unité motrice L'unité motrice est l'unité fonctionnelle de la contraction Les motoneurones α sont localisés dans la corne ventrale de la moelle épinière (ME) Leurs axones sortent de la moelle épinière en passant par la racine ventrale puis empruntent successivement les nerfs rachidiens et périphériques qui vont former de nombreuses ramifications allant innerver de multiples fibres musculaires (fm) Un seul motoneurone α peut innerver 35 (un des muscles extra-oculaires) à 2000 fm (ex le quadriceps) Chaque fm est innervée par un seul motoneurone α
 Leurs axones sortent de la moelle épinière en passant par la racine ventrale puis empruntent successivement les nerfs rachidiens et périphériques qui vont former de nombreuses ramifications allant innerver de multiples fibres musculaires (fm) Un seul motoneurone α peut innerver 35 (un des muscles extra-oculaires) à 2000 fm (ex le quadriceps) Chaque fm est innervée par un seul motoneurone α.")
4
Lien entre la nombre et le type d'unité motrice et son rôle
Le nombre de fibres musculaires contenues dans une unité motrice varie avec la taille du muscle et avec la finesse d'action de celui-ci : d'une dizaine pour le droit externe, petit muscle oculomoteur à 2000 environ pour le quadriceps fémoral. Ce nombre est ainsi lié au rôle fonctionnnel du muscle et à la capacité fonctionnelle de ces fibres, dont la contraction peut être lente ou rapide: les unités motrices de type lent comportent généralement un plus petit nombre de fibres musculaires que celles de type rapide. En outre, chaque unité motrice est composée de fibres d'un seul type.

6
Transmission de l’excitation à la fibre musculaire
Aspects électriques : les enregistrements intracellulaires montrent qu’en réponses à une stimulation électrique de l’axone, il se produit une dépolarisation de la plaque motrice. Lorsque ce potentiel de plaque motrice atteint une amplitude suffisante, il est le point de départ d’un potentiel propagé à l’ensemble de la fibre musculaire. Le délai qui sépare la stimulation de l’axone de l’apparition du potentiel propagé ajoute au temps de conduction le long de la partie distale de la fibre nerveuse un temps supplémentaire: délai synaptique.

7
Quand la somme algébrique des PPSE et des PPSI atteint un certain seuil au niveau du segment initial de l’axone du MN α les canaux sodiques sensibles au voltage s’ouvrent simultanément générant un potentiel d’action (PA) membranaire. Celui-ci se déplace le long de l’axone de manière saltatoire, sautant d’un noeud de Ranvier à l’autre Quand ce PA envahit les arborisations terminales,la dépolarisation de leur membrane ouvre les canaux calciques sensibles au voltage. L’entrée du calcium en leur sein conduit à la libération d’un neurotransmetteur, l’acétylcholine, dans la fente synaptique au niveau de la jonction neuromusculaire

8
Quand les molécules d’Ach se lient à leurs récepteurs, naissent les potentiels de plaques motrices miniatures:PPM (petites dépolarisation de la membrane muscul.), Simultanément les molécules d’Ach;sont métabolisées dans la fente synaptique par l’enzyme acetyl- cholinestérase Les PPM s’additionnent pour former un potentiel d’action au niveau de la mbrane de la fibre musculaire Ce PA envahi chaque fibre musc.par l’intermediaire du système de tubules T, constitué d’un ensemble de canaux perpendiculaires à l’axe longitudinal du muscle.(voir schéma) Lorsque le PA envahit le système de tubule T, de grandes quantés de CA sont libérées à partir du réticulum sarcoplasmique. Ce CA provoque successivement la formation et la rupture des liaisons existant entre l’actine et la myosine induisant ainsi le raccourcissement du muscle La somme intégrée au niv spatial de tous les PA des fm générée par la décharge d’un seul motoneurone alpha: le potentiel d’unité motrice (PUM)
 Lorsque le PA envahit le système de tubule T, de grandes quantés de CA sont libérées à partir du réticulum sarcoplasmique. Ce CA provoque successivement la formation et la rupture des liaisons existant entre l’actine et la myosine induisant ainsi le raccourcissement du muscle. La somme intégrée au niv spatial de tous les PA des fm générée par la décharge d’un seul motoneurone alpha: le potentiel d’unité motrice (PUM)")
10
Syndrome neurogène périphérique

11
Ι Définition: C’est l’ensemble de signes cliniques, éléctriques, biologiques et histologiques traduisant l’atteinte du neurone moteur périphérique, qu’il prenne ses origines dans les cornes antérieurs de la moelle ou dans les noyaux des nerfs crâniens et a n’import quel point de son trajet (plexus, racines, nerfs moteurs et sensitif).
.")
12
ΙΙ Rappel anatomo-physiologique
Les neurones alpha de type somatomoteur, sont situés dans les cornes antérieures de moelle, ou dans les noyaux des nerfs crâniennes; Chacun d’entre eux innerve un groupe de fibres musculaire et l’ensemble ainsi formé constituent une unité motrice;

13
Schéma d’un neurone Un motoneurone est un neurone qui transmet au muscle les signaux provenant du système nerveux central et périphérique. L’axone est un prolongement qui émerge du corps cellulaire du neurone. Il conduit l’influx nerveux sous la forme de signaux électriques. L’axone est recouvert par une gaine de myéline enveloppe isolante qui permet à l'influx nerveux de circuler plus rapidement. Cette gaine est faite d'une substance riche en lipides appelée la myéline.

14
Les nerfs rachidiens sont au nombre de 31 paires:
8 paires cervicales; 12 dorsales ; 5 lombaires; 5 sacrées ; 1 coccygienne. Les nerfs rachidiens sont constitués par l’union d’une racine antérieure (motrice) et d’une racine postérieure (sensitive), ils quittent le canal rachidien par le trou de conjugaison;
 et d’une racine postérieure (sensitive), ils quittent le canal rachidien par le trou de conjugaison;")
15
A leur sortie du trou de conjugaison les nerfs rachidiens se divisent en 02 branches:
Une branche postérieure la peau et aux muscles de la partie postérieure du corps Une branche antérieure téguments et des muscles de la partie antérieure du corps. Les branches antérieures s’anastomosent entre elles et forment les plexus troncs nerveux

16
ΙΙΙ- signes cliniques Déficit moteur: d’intensité variable selon le degré de l’atteinte, allant d’une simple diminution de la force musculaire à une paralysie complète et flasque; il est coté de 0 à 5; 0: absence de contraction volontaire 1: contraction faible sans déplacement 2: déplacement possible si l’action de la pesanteur est compensé 3: déplacement possible contre pesanteur 4: déplacement possible contre résistance 5: force musculaire normale.

17
La paralysie est complète, non dissociée, affectant la motilité volontaire, la motilité automatique et les réactions réflexes. On note une hypotonie de passivité et d’extensibilité. Topographie: elle est uni ou bilatérale, segmentaire ou diffuse, avec une prédominance à la racine ou à l’extrémité des membres (ailleurs on note une localisation distale).
.")
18
Amyotrophie : la conséquence de la dénervation du muscle.
Fasciculations: elles traduisent la mise en jeu d’une unité motrice dénervée, et se manifestent par des contractions (spontané ou provoqué) corne antérieure de la moelle.
 corne antérieure de la moelle.")
19
Contraction idiomusculaire: elle est normale
Les réflexes tendineux: sont abolis ou diminués, du fait de l’interruption de l’arc réflexe ; leur étude permet de déterminer le niveau de la lésion. La sensibilité: douleurs à type de radiculalgie ou névralgie; soit d’une atteinte de la sensibilité objective avec hypoesthésie de topographie radiculaire ou tronculaire; Ou atteinte de la sensibilité profonde grosses fibres myélinisés. Troubles vasomoteurs et trophiques: due à une atteinte des fibres du système nerveux autonomes Cyanose, œdème…

20
Examens complémentaires
EMG: il permet d’affirmer la nature neurogène périphérique du processus. L’électrodiagnostic de stimulation: Inexcitabilité ou hypo-excitabilité du muscle par le nerf La vitesse de conduction motrice est ralentie lorsqu’il s’agit d’une atteinte démyélinisante Réflexe H, onde F allongée. L’électrodiagnostic de détection : Au repos: activité électrique spontanée ( potentiel de fibrillation) qui traduit la contraction autonome des fibres musculaires énervés;
 qui traduit la contraction autonome des fibres musculaires énervés;")
21
Lors de la contraction volontaire: les résultats obtenus dépendent de l’importance des lésions; l’atteinte globale se traduit par un silence électrique, l’atteinte partielle se manifeste par un appauvrissement du tracé en potentiel d’unité motrice (Tracé pauvre) avec à l’extrême la présence d’une seule unité motrice ( Tracé simple). Il existe une sommation temporelle avec accélération du tracé.

22
Biopsie nerveuse: détermine le processus élémentaire de lésion (dégénérescence axonale ou démyélinisation segmentaire). Biopsie musculaire : atrophie musculaire avec systématisation fasciculaire.

23
Topographie-Étiologie
Atteinte du motoneurone alpha dans les cornes antérieures : déficit moteur, amyotrophie, fasciculation+++ : poliomyélites antérieures aigues ou chroniques Les amyotrophies spinales Sclérose latérale amyotrophique: syndrome pyramidal+ syndrome neurogène périphérique.

24
Atteinte radiculaire:
Signes subjectifs: douleurs de topographie fixe, continue avec des paroxysmes spontanées ou provoquées exacerbées par l’élévation du LCR (toux, éternuement, défécation) Signes objectifs: discret ou absent (anesthésie ou hypoesthésie). Les causes les plus fréquentes sont traumatique, envahissement néoplasique et la conséquence de la radiothérapie.
 Signes objectifs: discret ou absent (anesthésie ou hypoesthésie). Les causes les plus fréquentes sont traumatique, envahissement néoplasique et la conséquence de la radiothérapie.")
25
Atteintes plexiques : troubles sensitivo-moteurs sont localisés et unilatéraux, s’inscrivant dans le territoire de plusieurs racines superposées et adjacentes. Atteinte tronculaire : Mononeuropathie: la lésion intéresse un seul tronc nerveux expl: médian, cubital, ou radial… Mono neuropathie multiple: a caractère asynchrone et asymétrique expl : périartérite noueuse, diabète, porphyrie, amylose, infectieuse (lèpre) Polyneuropathie : d’installation habituellement progressive s’individualisent par le caractère symétrique et synchrone et la prédominance distale de l’atteinte sensitivo-motrice expl: cause
 Polyneuropathie : d’installation habituellement progressive s’individualisent par le caractère symétrique et synchrone et la prédominance distale de l’atteinte sensitivo-motrice expl: cause.")
26
carentielle : alcoolisme, plomb, arsenic.
cause infectieuse : diphtérie, fièvre de typhoïde, TBC cause métabolique : (diabète, insuffisance rénale) Polyradiculonévrite: atteinte conjointe des racines et des troncs nerveux s’associent à des troubles sensitifs; LCR: dissociation albumino- cytologique expl: Guillain et Barré.
 Polyradiculonévrite: atteinte conjointe des racines et des troncs nerveux s’associent à des troubles sensitifs; LCR: dissociation albumino- cytologique expl: Guillain et Barré.")
27
Polyneuropathie ascendante par dégénérescence
Représentation schématique des principales formes topographiques d’atteinte du système nerveux périphérique Polyneuropathie ascendante par dégénérescence rétrograde ("dying back")
")
28
Mononeuropathie et dégénérescence wallérienne distale

29
Neuropathie sensitive (ganglionopathie

30
Polyradiculonévrite (lésions de démyélinisation segmentaire réparties des racines aux parties distales)
")
31
PRINCIPAUX MÉCANISMES EN CAUSE DANS LES NEUROPATHIES PÉRIPHÉRIQUES ACQUISES

32
Processus pathologiques du nerf périphérique
Épinèvre F Périnèvre B Grosse fibre myélinisée Endonèvre A: Aspect Normal B: Perte axonale C: aspect de Demyélinisation avec bulbes d’oignons E; infiltrats cellulaires inflammatoire A C E D Fibre amyélinique

33
Electron micrograph: From Robert Schmidt MD
Late, large onion bulbs Abundant connective tissue around thinly myelinated axons. Onion bulbs may contain 0, 1, or several axons. Early onion bulb formations Note the few extra layers of basal lamina around several thinly myelinated larger axons Toluidine blue Large onion bulbs Several layers of basal lamina, connective tissue & Schwann cells around thinly myelinated axons

34
Syndrome Myopathique

35
Définition « c’est l’ensemble des manifestations cliniques, biologiques, électriques et histologique résultant de l’attente des muscles striés indépendamment de la commande nerveuse ou la jonction neuro- musculaire.

36
ΙΙ Rappel anatomo-physiologique

37
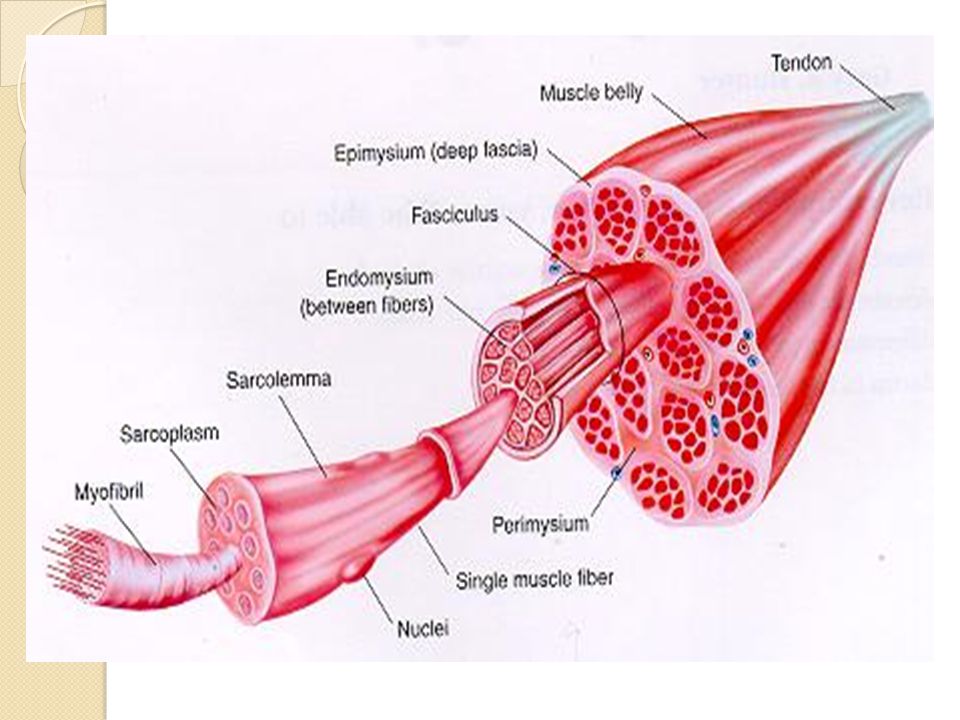
Anatomie du muscle strié squelettique Un muscle squelettique est entouré de plusieurs couches de tissu conjonctif : - l’endomysium entoure chaque fibre musculaire ; - le périmysium assemble les différentes fibres musculaires en faisceau de fibres musculaires ; - l’épimysium recouvre l’ensemble du muscle. Après avoir traversé l’épimysium, les vaisseaux sanguins (artérioles, veinules) qui assurent la vascularisation du muscle, donnent naissance à un fin réseau de capillaires qui gagne le périmysium puis l’endomysium pour vasculariser chaque fibre musculaire. Les prolongements des nerfs gagnent également le périmysium. Ils se terminent en arborisation dont les ramifications se terminent dans la jonction neuromusculaire pour innerver les différentes fibres musculaires.
 qui assurent la vascularisation du muscle, donnent naissance à un fin réseau de capillaires qui gagne le périmysium puis l’endomysium pour vasculariser chaque fibre musculaire. Les prolongements des nerfs gagnent également le périmysium. Ils se terminent en arborisation dont les ramifications se terminent dans la jonction neuromusculaire pour innerver les différentes fibres musculaires..")
39
De la fibre musculaire aux myofilaments De nombreuses myofibrilles occupent l’intérieur des fibres musculaires et en constituent les éléments contractiles. Les sarcomères se caractérisent par l’association, en une trame hexagonale, de filaments protéiques fins (actine) et épais (myosine). C’est le glissement des filaments les uns sur les autres qui réalise la contraction des myofibrilles.
 et épais (myosine). C’est le glissement des filaments les uns sur les autres qui réalise la contraction des myofibrilles..")
40
Membrane musculaire ou sarcolemme
LE SARCOLÈME RECOIT ET TRANSMET LE MESSAGE NERVEUX Le sarcolème est constitué d'une double couche de phospholipides, qui contient toutes sortes de protéines : protéines de structure, enzymes, canaux ioniques, récepteurs, transporteurs d'ions ou de molécules Les propriétés fonctionnelles du sarcolème dépendent de la nature de ces protéines, qui ne sont pas également réparties à la surface de la cellule. On distingue, en fait, deux régions : une région réceptrice de la commande nerveuse et une région conductrice.

41
Structure protéique de la membrane musculaire

42
Ι signes cliniques Déficit moteur:
Il constitue le symptôme essentiel du syndrome myopathique; Il en règle bilatéral et symétrique, à prédominance proximale; Selon le type de myopathie, le déficit peut affecter : Ceinture pelvienne: difficulté a se relever de la position accroupie ( signe de Gowers), impossibilité de se relever de la position assise (signe de Tabouret, démarche dandinante); Musculature rachidienne: trouble de la statique vertébrale avec en particulier hyperlordose lombaire; Ceinture scapulaire: difficulté à lever les bras au dessus de la tête, chute du moignon de l’épaule, décollement des omoplates; Les muscles de la face : faciès inexpressif, occlusion palpébrale incomplète; Muscles oculomoteurs : ophtalmoplégie
, impossibilité de se relever de la position assise (signe de Tabouret, démarche dandinante); Musculature rachidienne: trouble de la statique vertébrale avec en particulier hyperlordose lombaire; Ceinture scapulaire: difficulté à lever les bras au dessus de la tête, chute du moignon de l’épaule, décollement des omoplates; Les muscles de la face : faciès inexpressif, occlusion palpébrale incomplète; Muscles oculomoteurs : ophtalmoplégie.")
43
Ι signes cliniques Modification du volume musculaire:
Le plus souvent, Il s’agit d’une amyotrophie de topographie proximale et symétrique; Plus rarement, Il existe une pseudohypertrophie contrastant une diminution de la force musculaire; Abolition de la réponse idiomusculaire: la contraction musculaire normalement provoquée par la percussion du muscle est abolie.

44
Ι signes cliniques Autres signes: selon l’étiologie on peut parfois retrouver: Douleurs musculaires; Rétractions musculotendineuses; Une myotonie : retard et lenteur de la décontraction musculaire spontanée ou provoquée ( clinique , mécanique, électrique) Signes négatifs: Absence de trouble sensitif; Absence de signes centraux notamment pyramidaux; Conservation des réflexes ostéo-tendineux ( sauf dans les formes évolués).
 Signes négatifs: Absence de trouble sensitif; Absence de signes centraux notamment pyramidaux; Conservation des réflexes ostéo-tendineux ( sauf dans les formes évolués).")
45
ΙΙ examens complémentaires
Dosage des enzymes musculaires Les enzymes sont fréquemment élevés, de façon plus ou moins importantes selon l’étiologie; Les principales enzymes: La créatine phospho-kinase ( CPK); L’aldolase; La lactico-déshydrogénase (LDH); Les transaminases.
; L’aldolase; La lactico-déshydrogénase (LDH); Les transaminases.")
46
ΙΙ- examens complémentaires
EMG: lors de la contraction volontaire, l’EMG de détection montre un tracé de type myogène: trop riche et bas volté. Riche: il existe un recrutement spatial ( augmentation du nombre d’unités motrices recrutées ) Bas volté : les potentiels d’unités motrices ont une durée et une amplitude diminuées avec un aspect polyphasique. Au repos, l’EMG de détection peut retrouver une activité électrique anormale dans certaines pathologies : Salves myotoniques: potentiels battant a grande fréquence (120/sc) avec variation de fréquence et d’amplitude en début et fin de salve;
 Bas volté : les potentiels d’unités motrices ont une durée et une amplitude diminuées avec un aspect polyphasique. Au repos, l’EMG de détection peut retrouver une activité électrique anormale dans certaines pathologies : Salves myotoniques: potentiels battant a grande fréquence (120/sc) avec variation de fréquence et d’amplitude en début et fin de salve;")
47
ΙΙ examens complémentaires
Potentiels de fibrillation: traduisant l’excitabilité anormale des fibre musculaire (mypathie inflammatoires); Les vitesses de conduction nerveuse motrices et sensitives sont normales. Biopsie musculaire: Histologie avec colorations standards, en histochimie, en histoenzymologie et microscopie électronique; Aspect « Bariolé ».
; Les vitesses de conduction nerveuse motrices et sensitives sont normales. Biopsie musculaire: Histologie avec colorations standards, en histochimie, en histoenzymologie et microscopie électronique; Aspect « Bariolé ».")
48
ΙΙΙ Étiologies du syndrome myogène.
Affections musculaires acquises: Myopathies toxiques et iatrogènes: corticoïdes, colchicine, D- pénicillamine … Myopathies endocriennes; Myopathies inflammatoires. Dystrophies musculaires progressives: DMP avec myotonie (maladie de Steinert, maladie de Thomsen); DMP avec anomalie de la dystrophine (maladie de Duchenne, maladie de Becker); Myopathie facio-scapulo-humérale de Landouzy-Déjerine et autres DMP.
; DMP avec anomalie de la dystrophine (maladie de Duchenne, maladie de Becker); Myopathie facio-scapulo-humérale de Landouzy-Déjerine. et autres DMP.")
49
ΙΙΙ Étiologies du syndrome myogène.
Myopathies congénitales Début au cours de la période fœtale, hypotonie néonatale et déficit moteur; Biopsie musculaire, permet de distinguer différents types: a bâtonnets, central-core, centronucliaire; Myopathies métaboliques: Glycogénoses (perturbation du métabolisme des sucres); Lipidoses (perturbation des métabolismes des lipides; Myopathie mitochondriales.
; Lipidoses (perturbation des métabolismes des lipides; Myopathie mitochondriales.")
50
Atteinte de la jonction neuromusculaire

51
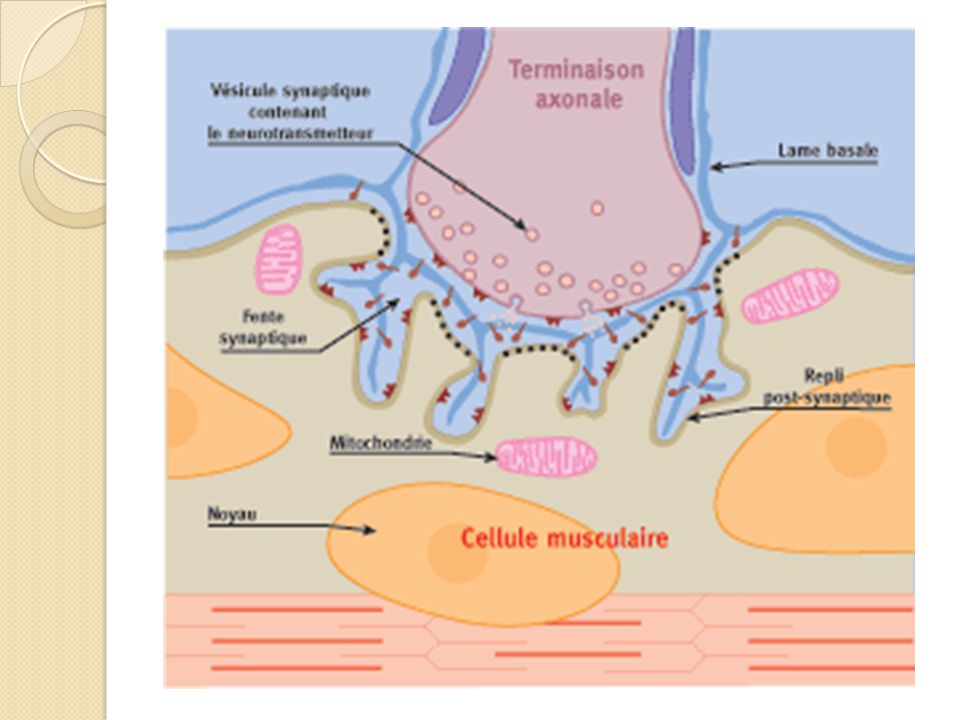
Ι. Rappel anatomo-physiologique
La jonction neuro-musculaire est constituée d’une portion présynaptique nerveuse et d’une portion postsynaptique musculaire

53
ΙΙ. Phénomène myasthénique
Le phénomène myasthénique est remarquable par la qualité du trouble moteur et par sa distribution; il s’agit de fatigabilité musculaire intermittentes n’existant pas au repos apparaissant lors d’une activation soutenue ou répétée du muscle revêtant un caractère progressif pouvant aboutir à une paralysie totale. il en résulte une distribution des troubles moteurs extrêmement variables d’un moment à l’autre. Le déficit myasthénique se marque électivement sur certains territoires.

54
Musculature oculaire :
diplopie transitoire et variable par atteinte des muscles oculo-moteurs; ptôsis asymétrique et male corrigé par le relèvement des sourcils; L’atteinte de la musculature oculaire est souvent bilatérale et non systématisée; Occlusion inefficace des paupières; La musculature intrinsèque de l’œil est toujours indemne et les réflexes pupillaires sont conservés.

55
Muscles de la face: aspect inexpressif de la mimique s’accentuant au cours de l’examen Impossibilité de siffler ou de souffler Déficit des muscles masticateurs s’accentuant au cours des repas au point de laisser une mâchoire tombante Muscles pharyngo-laryngés entraînant des troubles de la phonation et de la déglutition

56
Muscle des membres: le trouble myasthénique prédomine sur les muscles proximaux entraînant une difficulté pour se coiffer ( membres supérieurs) et des troubles de la marche ( membres inférieur)
 et des troubles de la marche ( membres inférieur)")
57
ΙΙΙ. Diagnostic Le diagnostic repose essentiellement sur trois éléments: Les caractères cliniques Quel soit le mode de début et la localisation de l'atteinte musculaire, certains caractères généraux permettent en effet de la rapportées à la myasthénie. Ces paralysies sans variables dans le temps et notamment elles sont plus marquées le soir que le matin. elle ne peuvent être analysée en terme de nerf mais seulement en terme de muscle. Elles sont isolées : en dehors des déficits moteurs, l'examen neurologique est tout à fait normal, notamment les réflexes ostéo-tendineux sont normaux, il n'y a pas de troubles sensitifs. La réaction de Mary Walker est positive.

58
Examen électrique: L’EMG de détection est peu significatif montrant des anomalies de types myogènes L’EMG de stimulation (répétitive) révèle l’existence d’un bloc myasténique. Test pharmacologique : confirme la nature du bloc L’injection en IV de 0.5 mg de Prostigmine ou Tensilon (10 mg) améliore temporairement la symptomatologie clinique Anticorps antirecepteur d’ACcholine
 révèle l’existence d’un bloc myasténique. Test pharmacologique : confirme la nature du bloc. L’injection en IV de 0.5 mg de Prostigmine ou Tensilon (10 mg) améliore temporairement la symptomatologie clinique. Anticorps antirecepteur d’ACcholine.")
59
Formes étiologiques Myasthénie auto-immune: Syndrome myasthénique:
Médicamenteux: expl:D-pénicillamine, colistine, gentamycine… Syndrome myasthénique par blocs présynaptique: Botulisme: Nausées, vomissement, troubles de la vision, perte de l’accommodation, mydriase aréactive EMG: potentialisation de la réponse lors de la stimulation répétitive à haute fréquence (30-90 Hz)
")
60
Syndrome de Lambert-Eaton:
Carcinomes pulmonaires a petites cellules Fatigabilité avec augmentation relative de la force musculaire EMG: potentialisation de réponse (30-47 Hz)
")
61
Stimulation à 3/s

Présentations similaires






. Il s’agit ici de nerfs rachidiens. A distinguer.>")
>")






>")






