Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
BIOINFORMATIQUE La bioinformatique : stocker, analyser et visualiser pour découvrir L’exemple du séquençage d’un génome Les banques de données Les banques de séquences nucléiques et protéiques Les banques d’alignements, de motifs et de sites La recherche dans les banques de données Analyser l’information La comparaison de séquences l’alignement multiple la phylogénie

2
I La bioinformatique : stocker, analyser et visualiser pour découvrir
Les progrès de la biotechnologie permettent aux chercheurs d’accéder à la séquence de plus en plus de gènes ou même de génomes complets. Chaque année, le nombre de nouvelles séquences double. Des systèmes efficaces de stockage de l’information doivent être mis en œuvre.

3
I La bioinformatique : stocker et analyser pour découvrir

4
I La bioinformatique : stocker et analyser pour découvrir
La production de ces séquences se fait de plus en plus dans le cadre de séquençages de génomes complets ou de banques d’EST (Expressed Sequence Tag) L’Homme (Homo sapiens) La mouche du vinaigre (Drosophila melanogaster) Un ver (Coenorhabditis elegans) Une plante (Arabidopsis thaliana) La levure (Saccharomyces cerevisiae) Une bactérie lactique (Bacillus subtilis) …
 L’Homme (Homo sapiens) La mouche du vinaigre (Drosophila melanogaster) Un ver (Coenorhabditis elegans) Une plante (Arabidopsis thaliana) La levure (Saccharomyces cerevisiae) Une bactérie lactique (Bacillus subtilis) …")
5
I La bioinformatique : stocker et analyser pour découvrir
Le séquençage des génomes est une tache complexe et gigantesque qui réclame la mise au point de logiciels capables d’automatiser la plupart des étapes Exemple du séquençage d’un génome complet I Production d’une banque BAC (Bacterial Artificial Chromosome) II Ordonnancement des BAC III Séquençage des BAC en « shot gun » IV Assemblage des séquences de BAC en chromosome V Annotation de la séquence du génome
 II Ordonnancement des BAC. III Séquençage des BAC en « shot gun » IV Assemblage des séquences de BAC en chromosome. V Annotation de la séquence du génome.")
6
Construction d’une banque BAC
chromosome Fragments chromosomiques BAC Banque BAC Inserts : ~ kpb

7
Ordonnancement de la banque BAC
Plusieurs méthodes - séquençage des extrémités - « fingerprinting » 1 2 4 6 5 3

8
Technique du « fingerprinting »
Analyse du profil de restriction des BAC BAC 1 BAC 2 Logiciels (Sanger Centre): IMAGE : lecture des gels FCP : assemblage
: IMAGE : lecture des gels. FCP : assemblage.")
9
Ordonnancement de la banque BAC
Plusieurs méthodes - séquençage des extrémités - « fingerprinting » 3 1 6 5 4 2

10
Séquençage « shot gun » Visualisation et édition des chromatogrammes
1 Amorces PCR aléatoires Visualisation et édition des chromatogrammes « base calling »

11
Lecture et nettoyage des séquences : PHRED
lecture des chromatogrammes élimination des bases de mauvaise qualité élimination des séquences contaminantes (BAC) assemblage des séquences : PHRAP, CAP3 recherche des séquences chevauchantes production d’une séquence consensus séquence consensus (contig)
 assemblage des séquences : PHRAP, CAP3. recherche des séquences chevauchantes. production d’une séquence consensus. séquence consensus (contig)")
12
Les difficultés de l’assemblage
Présence de séquences répétées Assemblage erroné CAP3 : règles et contraintes Intervention humaine

13
Banques et bases de données
annotations prédiction de gènes : intelligence artificielle (Eugène,…) prédiction de la fonction des gènes : comparaison de séquences (BLAST) Annotation structurale invertase I inconnu Annotation fonctionnelle Banques et bases de données
 prédiction de la fonction des gènes : comparaison de séquences (BLAST) Annotation structurale. invertase I. inconnu. Annotation fonctionnelle. Banques et bases de données.")
14
I La bioinformatique : stocker et analyser pour découvrir
La bioinformatique a pour objet de mettre en œuvre les moyens informatiques (bases de données, algorithmes) utiles au stockage et à l’analyse des données biologiques
 utiles au stockage et à l’analyse des données biologiques.")
15
I La bioinformatique : stocker et analyser pour découvrir
………………gaaa gaaactcgaa aacgagaaaa aaccatggcg aagtctgacg ctctcttgcc aatctccgcc agagaagaag atcctctatt atccgacggg tcaagatccg acccaaatgc cgaaacccat ggccgtagaa gacccgtgaa aggtctcctc gccgtctcat ttgggctttt ctttatcgcc ttctacgtcg ctctcatcgc cacacacgac ggatctagat ccaacgacgt taagatcgaa agcgatggaa cagcgaccaa agcgtcacgt gcccatctcg ccggcgtctc ggagaaaagc aatgatcagt tgtggaagct ttccggtgac aggaatacgg tggcgttctc atggaa………… …………… cgaa tccgaatgca gatggacagt agacattgca gatacccaga ttagacaggg tgtaagcgat ggaaatgaac agattgacag tagacaggat aacaagatac cagctcgata Cagataccgc tagacataga caccatgcag atgttcatta gataccagat agggacagat Gacagataga ggacatagcg ctgcgtacac agatactcgg ataggacata tatagacaga Cagatataga ctcagataga cgctcgacat cgctagacag ctctcgccgt gcatagacca Gatgacagat ggcgtgcgtc gtagtactgc atcgtcatcg aatgccggta ttcgatcgaa Cgtgca………… L’une de ses séquences est artificielle. Laquelle ?

16
I La bioinformatique : stocker et analyser pour découvrir
La composition des génomes Le rapport (G+C)/(A+T) ou le pourcentage de GC Escherichia coli 51 % Plasmodium falciparum 18 % Thermus thermophilus 68 % Vertébrés % (et présence d’isochores) Le pourcentage de GC varie entre 15 et 75 %. L’origine de ces variations est encore mal comprise.
/(A+T) ou le pourcentage de GC. Escherichia coli 51 % Plasmodium falciparum 18 % Thermus thermophilus 68 % Vertébrés 40-45% (et présence d’isochores) Le pourcentage de GC varie entre 15 et 75 %. L’origine de ces variations est encore mal comprise.")
17
I La bioinformatique : stocker et analyser pour découvrir
La composition des protéines Relativement constante a travers le vivant. Alanine (A) ‰ Cysteine (C) ‰ Méthionine (M) 24‰ Asparagine (N) 44‰ Proline (P) ‰ Aspartate (D) ‰ Glutamate (E) ‰ Glutamine (Q) ‰ Phénylalanine (F) 39‰ Arginine (R) ‰ Glycine (G) ‰ Sérine (S) ‰ Histidine (H) ‰ Thréonine (T) ‰ Isoleucine (I) ‰ Valine (V) ‰ Lysine (K) % Tryptophane (W) 13‰ Leucine (L) ‰ Tyrosine (Y) ‰ Cette distribution moyenne peut être considérée comme une signature
 83‰ Cysteine (C) 17‰ Méthionine (M) 24‰ Asparagine (N) 44‰ Proline (P) 51‰ Aspartate (D) 53‰ Glutamate (E) 62‰ Glutamine (Q) 40‰ Phénylalanine (F) 39‰ Arginine (R) 57‰ Glycine (G) 72‰ Sérine (S) 69‰ Histidine (H) 22‰ Thréonine (T) 58‰ Isoleucine (I) 52‰ Valine (V) 66‰ Lysine (K) 57% Tryptophane (W) 13‰ Leucine (L) 90‰ Tyrosine (Y) 32‰ Cette distribution moyenne peut être considérée comme une signature.")
18
I La bioinformatique : stocker et analyser pour découvrir
Etude des fréquences n-uplets Comparaison de la fréquence d’apparition d’un n-uplet fB1B2…Bn au produit des fréquences d’apparition des bases individuelles fB1.fB2….fBn Si fB1B2…Bn > fB1.fB2….fBn le n-uplet est sur-représenté Si fB1B2…Bn < fB1.fB2….fBn le n-uplet est sous-représenté Exemple : Chez E. coli, f CTAG = 3, << fCfTfAfG = 3,9 10-3 Les palindromes sont en général sous-représentés dans les génomes bactériens, les palindromes sont souvent des sites de restriction. Chez les vertébrés, le dinucléotide GC est rare. Il s’agit d’un signal de méthylation de la cytosine. La 5-méthyl-cytosine peut ensuite être transformée en T. Ainsi CG se raréfie au profit de TG.

19
I La bioinformatique : stocker et analyser pour découvrir
Un n-uplet particulier le codon La distribution des codons doit suivre celle des acides aminés qui leurs correspondent dans les protéines ainsi : fW = fTGG Etude de l’usage des codons synonymes Il existe donc des codons privilégiés. Ces codons ne sont pas les mêmes d’une espèce à l’autre. lysine E. Coli H. Sapiens AAA 60 % 38 % AAG 40 % 62 %

20
I La bioinformatique : stocker et analyser pour découvrir
En étudiant un grand nombre de gènes on peut construire, pour l’organisme auquel ces gènes appartiennent une table d’usage des codons. Cette table diffère de celles construites pour d’autre organismes, mais on observe une conservation évolutive : des espèces proches possèdent des tables d’usage des codons proches.

21
I La bioinformatique : stocker et analyser pour découvrir
Un n-uplet particulier le codon Homo sapiens [gbpri]: CDS's ( codons) fields: [triplet] [amino acid] [fraction] [frequency: per thousand] ([number]) UUU F (374332) UCU S (323470) UAU Y (264652) UGU C (221863) UUC F (448127) UCC S (384476) UAC Y (339473) UGC C (271056) UUA L (160731) UCA S (260418) UAA * ( 16884) UGA * ( 30111) UUG L (277774) UCG S ( 98166) UAG * ( 12911) UGG W (284246) CUU L (283480) CCU P (380219) CAU H (231860) CGU R (102673) CUC L (428574) CCC P (439256) CAC H (329569) CGC R (236986) CUA L (153837) CCA P (367297) CAA Q (261063) CGA R (138297) CUG L (880072) CCG P (154028) CAG Q (755209) CGG R (257761) AUU I (346233) ACU T (283671) AAU N (365457) AGU S (263279) AUC I (466577) ACC T (419213) AAC N (422697) AGC S (424788) AUA I (157385) ACA T (325763) AAA K (526117) AGA R (255681) AUG M (489160) ACG T (135294) AAG K (713826) AGG R (254743) GUU V (239795) GCU A (408931) GAU D (484271) GGU G (237026) GUC V (320190) GCC A (622538) GAC D (563848) GGC G (495700) GUA V (154102) GCA A (350382) GAA E (634985) GGA G (358824) GUG V (630151) GCG A (165700) GAG E (884368) GGG G (360728) Coding GC 52.58% 1st letter GC 56.14% 2nd letter GC 42.46% 3rd letter GC 59.13% Genetic code 1: Standard
 fields: [triplet] [amino acid] [fraction] [frequency: per thousand] ([number]) UUU F (374332) UCU S (323470) UAU Y (264652) UGU C (221863) UUC F (448127) UCC S (384476) UAC Y (339473) UGC C (271056) UUA L (160731) UCA S (260418) UAA * ( 16884) UGA * ( 30111) UUG L (277774) UCG S ( 98166) UAG * ( 12911) UGG W (284246) CUU L (283480) CCU P (380219) CAU H (231860) CGU R (102673) CUC L (428574) CCC P (439256) CAC H (329569) CGC R (236986) CUA L (153837) CCA P (367297) CAA Q (261063) CGA R (138297) CUG L (880072) CCG P (154028) CAG Q (755209) CGG R (257761) AUU I (346233) ACU T (283671) AAU N (365457) AGU S (263279) AUC I (466577) ACC T (419213) AAC N (422697) AGC S (424788) AUA I (157385) ACA T (325763) AAA K (526117) AGA R (255681) AUG M (489160) ACG T (135294) AAG K (713826) AGG R (254743) GUU V (239795) GCU A (408931) GAU D (484271) GGU G (237026) GUC V (320190) GCC A (622538) GAC D (563848) GGC G (495700) GUA V (154102) GCA A (350382) GAA E (634985) GGA G (358824) GUG V (630151) GCG A (165700) GAG E (884368) GGG G (360728) Coding GC 52.58% 1st letter GC 56.14% 2nd letter GC 42.46% 3rd letter GC 59.13% Genetic code 1: Standard.")
22
I La bioinformatique : stocker et analyser pour découvrir
H.sapiens UGG W A thaliana UGG W T aquaticus UGG W H. sapiens GGU G GGC G GGA G GGG G A. thaliana GGU G GGC G GGA G GGG G T. Aquaticus GGU G GGC G GGA G GGG G

23
I La bioinformatique : stocker et analyser pour découvrir
Effet de la composition en base du génome sur l’usage des codons : Les organismes riches en GC auront une préférence significative pour les codons possédant un G ou un C comme troisième base. C’est l’inverse pour les organismes riches en AT Pour les autres organismes, le choix de la troisième base reste fortement biaisé.

24
I La bioinformatique : stocker et analyser pour découvrir
L’effet de contexte : Si deux codons synonymes ont un usage proche, alors le choix peut être influencé par le contexte, c’est à dire par les nucléotides présents immédiatement en amont ou en aval du codon. Exemple : Chez E. coli, pour la lysine, on trouve plus fréquemment AAA lorsque le codon suivant commence par G et AAG est préféré si un C est le nucléotide en aval.

25
I La bioinformatique : stocker et analyser pour découvrir
L’usage des codons et l’expression des gènes. Chez la levure (Saccharomyces cerevisiae) et E. coli, la fréquence d’usage des codons est directement proportionnelle à la concentration cellulaire de l’ARNt correspondant. Il s’agit d’une adaptation qui permet d’ajuster la quantité d’ARNt aux besoins de la machinerie de biosynthèse protéique. Les gènes le plus exprimés sont ceux qui utilisent le plus de codons privilégiés. L’utilisation de codons rares permet d’introduire des poses dans la traduction.
 et E. coli, la fréquence d’usage des codons est directement proportionnelle à la concentration cellulaire de l’ARNt correspondant. Il s’agit d’une adaptation qui permet d’ajuster la quantité d’ARNt aux besoins de la machinerie de biosynthèse protéique. Les gènes le plus exprimés sont ceux qui utilisent le plus de codons privilégiés. L’utilisation de codons rares permet d’introduire des poses dans la traduction.")
26
I La bioinformatique : stocker et analyser pour découvrir
Ces résultats statistiques peuvent permettre d’analyser les nouvelles séquences pour rechercher les phases codantes, les limites intron/exon, les erreurs de séquençage. Tous ces éléments combinés permettent de prédire la position de gènes. C’est l’annotation structurale. Prediction des zones introniques et exoniques au moyen de méthodes statistiques. Recherche des motifs accepteurs et donneurs d’épissage Combinaison des deux infos précédentes pour prédire précisément les limites des introns/exons Assemblage des exons prédits et confrontation avec les banques d’EST de l’organisme considéré Si échec, confronter les protéines prédites aux protéines existantes dans les bases de données (pour d’autres organismes) Il existe des logiciels qui combinent toutes ces approches tel GenScan qui a été utilisé lors du séquençage du génome humain
 Il existe des logiciels qui combinent toutes ces approches tel GenScan qui a été utilisé lors du séquençage du génome humain.")
27
II Les banques de données
L’ensemble des séquences nucléiques ou protéiques connues sont regroupées dans des banques de données GENBANK au NCBI (National Centre for Biotechnology Information, USA) EMBL à l’EBI (European Molecular Biology Laboratory, European Bioinformatics Institute, UK) DDBJ au Japon (DNA Data Bank of Japan) Certaines banques ne contiennent que des séquences protéiques UNIPROT (Swissprot) à l’ISB/EBI (Institut Suisse de bioinformatique) PIR , Georgetown University, USA (Protein Information Resource)
 EMBL à l’EBI (European Molecular Biology Laboratory, European Bioinformatics Institute, UK) DDBJ au Japon (DNA Data Bank of Japan) Certaines banques ne contiennent que des séquences protéiques. UNIPROT (Swissprot) à l’ISB/EBI (Institut Suisse de bioinformatique) PIR , Georgetown University, USA (Protein Information Resource)")
28
II Les banques de données
II.1 L’organisation de l’information : Banque de données : l’information est stockée sous la forme d’une collection de fichiers structurés. Une séquence correspond à un fichier. Base de données : l’information est stockée dans les champs d’un SGBD (Système de Gestion de Base de Données). Un langage particulier permet de formuler des requêtes pour interroger la base (SQL, Structured Query Langage)
. Un langage particulier permet de formuler des requêtes pour interroger la base (SQL, Structured Query Langage)")
29
II Les banques de données
II.2 Les banques de séquences nucléiques GENBANK, EMBL et DDBJ sont associées et diffusent les mêmes informations, mais sous des formats légèrement différents. Ces banques sont toutes accessibles via Internet à quiconque et sans restriction Elles gèrent les plus de 10 millions de séquences connues à ce jour, quel que soit leur organisme d’origine

30
II Les banques de données
II.2.a GENBANK Les séquences sont pour une large part obtenues par soumission directe des chercheurs ou via les grands programmes de séquençage. Pour simplifier les recherches des utilisateurs, Genbank, EMBL et DDBJ s’échangent régulièrement leurs données de sorte que les trois banques disposent en permanence des mêmes séquences Par commodité les séquences sont classées en divisions selon leur type (EST, séquençage massif,…) ou leur organisme d’origine. Il existe une vingtaine de ces divisions
 ou leur organisme d’origine. Il existe une vingtaine de ces divisions.")
31
II Les banques de données
II.2.a GENBANK DIVISIONS Utilisées par quelles banques ? BCT Bacteries DDBJ, GenBank PRO Procaryotes EMBL FUN Champignons EMBL HUM Humain DDBJ, EMBL PRI Primates DDBJ, EMBL, GenBank ROD Rongeurs DDBJ, EMBL, GenBank MAM Autre mammifères DDBJ, EMBL, GenBank VRT Autres vertébrés DDBJ, EMBL, GenBank INV Invertébrés DDBJ, EMBL, GenBank PLN Plantes DDBJ, EMBL, GenBank ORG Organelles EMBL VRL Virus DDBJ, EMBL, GenBank PHG Phages DDBJ, EMBL, GenBank RNA ARN de tructure DDBJ, EMBL, GenBank SYN Synthétiques et chimériques DDBJ, EMBL, GenBank UNA Non annotées DDBJ, GenBank UNC Non classifiées EMBL

32
II Les banques de données
II.2.a GENBANK Divisions fonctionnelles Utilisées par quelles banques ? EST Expressed sequence tags DDBJ, EMBL, GenBank STS Sequence tagged sites DDBJ, EMBL, GenBank GSS Genome survey sequences DDBJ, EMBL, GenBank HTG High throughput genomic sequences DDBJ, EMBL, GenBank PAT Patent sequences DDBJ, EMBL, GenBank CON* Virtual contigs of segmented sequences DDBJ, EMBL, GenBank

33
II Les banques de données
II.2.a GENBANK Chaque séquence possède une « entrée » qui rassemble toute l’information la concernant. Cette information peut-être visualisée sous forme d’une « fiche » Exemple : l’ARNm de l’invertase acide de Brassica oleracea

34
II Les banques de données
II.2.a GENBANK LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand.")
35
II Les banques de données
II.2.a GENBANK Nom de la séquence taille molécule division LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand.")
36
II Les banques de données
II.2.a GENBANK LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand Description de la séquence : origine, type, fonction, … Champ texte libre
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand. Description de la séquence : origine, type, fonction, … Champ texte libre.")
37
II Les banques de données
II.2.a GENBANK LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand Numéro d’accession primaire. Ne change jamais. En cas de fusion ou de scission d’une entrée, des numéros secondaires peuvent apparaître.
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand. Numéro d’accession primaire. Ne change jamais. En cas de fusion ou de scission. d’une entrée, des numéros secondaires peuvent. apparaître.")
38
II Les banques de données
II.2.a GENBANK LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand Numéro de version. Il est incrémenté à chaque modification. Seule la dernière version est accessible directement. Le deuxième numéro GI permet de satisfaire à des contraintes techniques.
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand. Numéro de version. Il est incrémenté à chaque modification. Seule la dernière version est accessible directement. Le deuxième numéro GI permet de satisfaire à des contraintes. techniques.")
39
II Les banques de données
II.2.a GENBANK LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand Mots-clés : un mot ou une courte phrase, Les mots-clés sont séparés par une « , » Les mots-clés sont librement choisis par les auteurs.
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand. Mots-clés : un mot ou une courte phrase, Les mots-clés sont séparés par une « , » Les mots-clés sont librement choisis par les auteurs.")
40
II Les banques de données
II.2.a GENBANK LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand Organisme et éventuellement type de molécule
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand. Organisme et éventuellement type de molécule.")
41
II Les banques de données
II.2.a GENBANK LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand.")
42
II Les banques de données
II.2.a GENBANK LOCUS AF bp mRNA PLN NOV-2001 DEFINITION Brassica oleracea clone BoINV2 acid invertase mRNA, complete cds. ACCESSION AF274299 VERSION AF GI: KEYWORDS . SOURCE Brassica oleracea. ORGANISM Brassica oleracea Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; eurosids II; Brassicales; Brassicaceae; Brassica. REFERENCE 1 (bases 1 to 2251) AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during senescence of broccoli (Brassica oleracea) florets JOURNAL Unpublished REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand
 AUTHORS Coupe,S.A., Sinclair,B.K., Greer,L. and Hurst,P.L. TITLE Characterization of acid invertase gene expression during. senescence of broccoli (Brassica oleracea) florets. JOURNAL Unpublished. REFERENCE 2 (bases 1 to 2251) AUTHORS Coupe,S.A. TITLE Direct Submission. JOURNAL Submitted (02-JUN-2000) Crop and Food Research, Private Bag , Palmerston North, Manawatu 5301, New Zealand.")
43
FEATURES Location/Qualifiers
source /organism="Brassica oleracea" /cultivar="Shogun" /db_xref="taxon:3712" /clone="BoINV2" CDS /EC_number=" " /function="cleaves sucrose into glucose and fructose at acid pH optima" /note="sucrose hydrolysing enzyme; beta-fructofuranosidase" /codon_start=1 /product="acid invertase" /protein_id="AAG " /db_xref="GI: " /translation="MAKSDALLPISAREEDPLLSDGSRSDPNAETHGRRRPVKGLLAV SFGLFFIAFYVALIATHDGSRSNDVKIESDGTATKASRAHLAGVSEKSNDQLWKLSGD RNTVAFSWNNSMLSWQRTAFHFQPEQNWMNDPNGPLFYKGWYHFFYQYNPNAAVWGDI VWGHAVSKDLIHWVHLPLAMAADQWYDANGVWTGSATFLEDGSIVMLYTGSTDKSVQV QNLAYPEDLNDPLLLKWVKFPGNPVLVPPPGILPKDFRDPTTAWKTSAGKWRITIGSK INRTGISLVYDTTDFKTYEKLETLLHKVPNTGMWECVDFYPVSKTLVKGLDTSVNGPD VKHIVKASMDDTRIDHYAIGTYFDSNGTWTPDDPTIDVGISTSLRYDYGKFYASKTFY DQNKGRRILWGWIGESDSESADVQKGWSSLQGIPRTVVLDTKTGKNLVQWPVEEVKSL RLSSKKFDMEVGPGSLVHIDVGSAAQLDIEAEFEIKKESLEKILGDASAAAEAEEFSC QKSGGSTVRGALGPFGFSVLAHESLSEKTPVYFYVAKGKDSKLITFFCTDSSRSSFAN DVVKPIYGSSVPVLKGEKLTMRILVDHSIVEAFGQGGRTCITSRVYPTKAIYGAAKLF LFNNAIDATITASFKVWQMNSAFIQPYSEEAVRALSRT"

44
FEATURES Location/Qualifiers Table des « features » :
source /organism="Brassica oleracea" /cultivar="Shogun" /db_xref="taxon:3712" /clone="BoINV2" CDS /EC_number=" " /function="cleaves sucrose into glucose and fructose at acid pH optima" /note="sucrose hydrolysing enzyme; beta-fructofuranosidase" /codon_start=1 /product="acid invertase" /protein_id="AAG " /db_xref="GI: " /translation="MAKSDALLPISAREEDPLLSDGSRSDPNAETHGRRRPVKGLLAV SFGLFFIAFYVALIATHDGSRSNDVKIESDGTATKASRAHLAGVSEKSNDQLWKLSGD RNTVAFSWNNSMLSWQRTAFHFQPEQNWMNDPNGPLFYKGWYHFFYQYNPNAAVWGDI VWGHAVSKDLIHWVHLPLAMAADQWYDANGVWTGSATFLEDGSIVMLYTGSTDKSVQV QNLAYPEDLNDPLLLKWVKFPGNPVLVPPPGILPKDFRDPTTAWKTSAGKWRITIGSK INRTGISLVYDTTDFKTYEKLETLLHKVPNTGMWECVDFYPVSKTLVKGLDTSVNGPD VKHIVKASMDDTRIDHYAIGTYFDSNGTWTPDDPTIDVGISTSLRYDYGKFYASKTFY DQNKGRRILWGWIGESDSESADVQKGWSSLQGIPRTVVLDTKTGKNLVQWPVEEVKSL RLSSKKFDMEVGPGSLVHIDVGSAAQLDIEAEFEIKKESLEKILGDASAAAEAEEFSC QKSGGSTVRGALGPFGFSVLAHESLSEKTPVYFYVAKGKDSKLITFFCTDSSRSSFAN DVVKPIYGSSVPVLKGEKLTMRILVDHSIVEAFGQGGRTCITSRVYPTKAIYGAAKLF LFNNAIDATITASFKVWQMNSAFIQPYSEEAVRALSRT" Table des « features » : Mis en place par GenBank, EMBL et DDBJ Contient des informations sur les gènes et leurs produits ainsi que sur les régions d’intérêt biologique des séquences. On y trouve aussi des informations sur les différences entre les versions d’une même séquence. Des liens sur d’autres bases ou banques de données peuvent également être présents. Chaque clé répond à une nomenclature.

45
Lien vers la base Taxon du NCBI
FEATURES Location/Qualifiers source /organism="Brassica oleracea" /cultivar="Shogun" /db_xref="taxon:3712" /clone="BoINV2" CDS /EC_number=" " /function="cleaves sucrose into glucose and fructose at acid pH optima" /note="sucrose hydrolysing enzyme; beta-fructofuranosidase" /codon_start=1 /product="acid invertase" /protein_id="AAG " /db_xref="GI: " /translation="MAKSDALLPISAREEDPLLSDGSRSDPNAETHGRRRPVKGLLAV SFGLFFIAFYVALIATHDGSRSNDVKIESDGTATKASRAHLAGVSEKSNDQLWKLSGD RNTVAFSWNNSMLSWQRTAFHFQPEQNWMNDPNGPLFYKGWYHFFYQYNPNAAVWGDI VWGHAVSKDLIHWVHLPLAMAADQWYDANGVWTGSATFLEDGSIVMLYTGSTDKSVQV QNLAYPEDLNDPLLLKWVKFPGNPVLVPPPGILPKDFRDPTTAWKTSAGKWRITIGSK INRTGISLVYDTTDFKTYEKLETLLHKVPNTGMWECVDFYPVSKTLVKGLDTSVNGPD VKHIVKASMDDTRIDHYAIGTYFDSNGTWTPDDPTIDVGISTSLRYDYGKFYASKTFY DQNKGRRILWGWIGESDSESADVQKGWSSLQGIPRTVVLDTKTGKNLVQWPVEEVKSL RLSSKKFDMEVGPGSLVHIDVGSAAQLDIEAEFEIKKESLEKILGDASAAAEAEEFSC QKSGGSTVRGALGPFGFSVLAHESLSEKTPVYFYVAKGKDSKLITFFCTDSSRSSFAN DVVKPIYGSSVPVLKGEKLTMRILVDHSIVEAFGQGGRTCITSRVYPTKAIYGAAKLF LFNNAIDATITASFKVWQMNSAFIQPYSEEAVRALSRT" Lien vers la base Taxon du NCBI Lien vers la base Enzyme Lien vers la fiche de la protéine dans Genbank

46
II Les banques de données
II.2.a GENBANK BASE COUNT a c g t ORIGIN 1 caaaaagaaa gaaactcgaa aacgagaaaa aaccatggcg aagtctgacg ctctcttgcc 61 aatctccgcc agagaagaag atcctctatt atccgacggg tcaagatccg acccaaatgc 121 cgaaacccat ggccgtagaa gacccgtgaa aggtctcctc gccgtctcat ttgggctttt 181 ctttatcgcc ttctacgtcg ctctcatcgc cacacacgac ggatctagat ccaacgacgt 241 taagatcgaa agcgatggaa cagcgaccaa agcgtcacgt gcccatctcg ccggcgtctc 301 ggagaaaagc aatgatcagt tgtggaagct ttccggtgac aggaatacgg tggcgttctc 361 atggaacaac agtatgttgt cgtggcaacg aacggcgttt catttccaac ctgaacagaa 421 ctggatgaac gatcctaatg gtccattgtt ctacaaagga tggtaccatt tcttctacca 481 gtacaaccca aacgcagcag tatggggtga cattgtttgg ggtcatgccg tgtctaagga /../ 1861 aagggtatat ccaacaaagg ccatctatgg agcagcgaag cttttcttgt tcaacaatgc 1921 cattgatgcg actattacgg catcgtttaa ggtgtggcag atgaacagtg cttttattca 1981 gccttactct gaggaggctg ttcgtgctct ctcccgcaca tgattataca cccatctcca 2041 gcaaattctt tttttttttt ttttgtagat ttacttatta aaacttataa atatcgttct 2101 gttattcttc caatttagct cgttcaatta ttctattggg gttcaatttg attcatcata 2161 tgtaagaaaa atgggttact tgagaaattt tttttctcat tatctttaat aaaattttgg 2221 tgaaaaaaaa aaaaaaaaaa aaaaaaaaaa a //

47
II Les banques de données
II.2.a GENBANK Contenu de Genbank

48
II Les banques de données
II.2.b Visualisation des séquences nucléiques EMBL et DDBJ stockent les même séquences sous un format très similaire Devant la complexité croissante des fiches et notamment avec l’arrivée des tables de « features », de nouveaux outils ont été développés pour visualiser ces fiches. ARTEMIS est l’un d’entre-eux Développé en langage JAVA (multiplateforme) Logiciel libre (gratuit) Visualise toute fiche GenBank/EMBL Permet d’éditer ces fiches et de créer de nouvelles annotations
 Logiciel libre (gratuit) Visualise toute fiche GenBank/EMBL. Permet d’éditer ces fiches et de créer de nouvelles annotations.")
50
II Les banques de données
II.3 Les banques de séquences protéiques PIR / NRL-3D PIR littérature, soumissions, traductions de Genbank, EMBL et DDBJ annotations automatiques, classification en familles (50% d’identité), superfamilles, domaines annotations bibliographiques et vérifications (PIR1 et PIR2) entrées classifiées mais d’annotation pauvre NRL-3D séquences et annotations issues de la PDB entrées
, superfamilles, domaines. annotations bibliographiques et vérifications (PIR1 et PIR2) entrées classifiées mais d’annotation pauvre. NRL-3D. séquences et annotations issues de la PDB entrées.")
51
II Les banques de données II.3 Les banques de séquences protéiques
II.3.a SWISS-PROT / TREMBL SWISS-PROT Origine des séquences : littérature, soumissions Annotations manuelles (littérature, experts) entrées (10/01) TREMBL Traduction des CDS de EMBL par le programme trembl ORF (Open Reading Frame) : Phase ouverte de lecture; séquence nucléique comprise entre deux codons stop CDS (coding sequence) : sequence nucléique codant pour une protéine. Elle est contenue dans une phase ouverte de lecture et débute par un codon start. Annotations automatiques SP-TREMBL entrées Après expertise les fiches TREMBL validée sont transférées dans SWISS-PROT
 entrées (10/01) TREMBL. Traduction des CDS de EMBL par le programme trembl. ORF (Open Reading Frame) : Phase ouverte de lecture; séquence nucléique comprise entre deux codons stop. CDS (coding sequence) : sequence nucléique codant pour une protéine. Elle est contenue dans une phase ouverte de lecture et débute par un codon start. Annotations automatiques SP-TREMBL entrées. Après expertise les fiches TREMBL validée sont transférées dans SWISS-PROT.")
52
Standard/preliminary
II.3.a SWISS-PROT / TREMBL ID line : toujours la première ligne, elle contient le nom de la séquence au format X_Y X = nom de la protéine ( mnémonique de 4 lettres) Y = code espèce (5 lettres genre (3) espèce (2)) Classe : Standard/preliminary taille ID HXK1_ARATH STANDARD; PRT; AA. AC Q42525; Q42535; DT 01-NOV-1997 (Rel. 35, Created) DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 16-OCT-2001 (Rel. 40, Last annotation update) DE Hexokinase 1 (EC ). GN HXK1 OR AT4G29130 OR F19B OS Arabidopsis thaliana (Mouse-ear cress). OC Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; OC Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; OC eurosids II; Brassicales; Brassicaceae; Arabidopsis. OX NCBI_TaxID=3702;
 Y = code espèce (5 lettres genre (3) espèce (2)) Classe : Standard/preliminary. taille. ID HXK1_ARATH STANDARD; PRT; 496 AA. AC Q42525; Q42535; DT 01-NOV-1997 (Rel. 35, Created) DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 16-OCT-2001 (Rel. 40, Last annotation update) DE Hexokinase 1 (EC ). GN HXK1 OR AT4G29130 OR F19B OS Arabidopsis thaliana (Mouse-ear cress). OC Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; OC Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; OC eurosids II; Brassicales; Brassicaceae; Arabidopsis. OX NCBI_TaxID=3702;")
53
II.3.a SWISS-PROT / TREMBL
ID HXK1_ARATH STANDARD; PRT; AA. AC Q42525; Q42535; DT 01-NOV-1997 (Rel. 35, Created) DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 16-OCT-2001 (Rel. 40, Last annotation update) DE Hexokinase 1 (EC ). GN HXK1 OR AT4G29130 OR F19B OS Arabidopsis thaliana (Mouse-ear cress). OC Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; OC Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; OC eurosids II; Brassicales; Brassicaceae; Arabidopsis. OX NCBI_TaxID=3702; Numéros d’accession primaire et secondaire(s)
 DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 16-OCT-2001 (Rel. 40, Last annotation update) DE Hexokinase 1 (EC ). GN HXK1 OR AT4G29130 OR F19B OS Arabidopsis thaliana (Mouse-ear cress). OC Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; OC Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; OC eurosids II; Brassicales; Brassicaceae; Arabidopsis. OX NCBI_TaxID=3702; Numéros d’accession primaire et secondaire(s)")
54
II.3.a SWISS-PROT / TREMBL
ID HXK1_ARATH STANDARD; PRT; AA. AC Q42525; Q42535; DT 01-NOV-1997 (Rel. 35, Created) DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 16-OCT-2001 (Rel. 40, Last annotation update) DE Hexokinase 1 (EC ). GN HXK1 OR AT4G29130 OR F19B OS Arabidopsis thaliana (Mouse-ear cress). OC Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; OC Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; OC eurosids II; Brassicales; Brassicaceae; Arabidopsis. OX NCBI_TaxID=3702; Description : champ texte
 DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 16-OCT-2001 (Rel. 40, Last annotation update) DE Hexokinase 1 (EC ). GN HXK1 OR AT4G29130 OR F19B OS Arabidopsis thaliana (Mouse-ear cress). OC Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; OC Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; OC eurosids II; Brassicales; Brassicaceae; Arabidopsis. OX NCBI_TaxID=3702; Description : champ texte.")
55
II.3.a SWISS-PROT / TREMBL
synonyme DE Corticotropin-lipotropin precursor (Pro-opiomelanocortin) (POMC) DE [Contains: NPP; Melanotropin gamma (Gamma-MSH); Corticotropin DE (Adrenocorticotropic hormone) (ACTH); Melanotropin alpha (Alpha-MSH); DE Corticotropin-like intermediary peptide (CLIP); Lipotropin beta (Beta- DE LPH); Lipotropin gamma (Gamma-LPH); Melanotropin beta (Beta-MSH); DE Beta-endorphin; Met-enkephalin].
 (POMC) DE [Contains: NPP; Melanotropin gamma (Gamma-MSH); Corticotropin. DE (Adrenocorticotropic hormone) (ACTH); Melanotropin alpha (Alpha-MSH); DE Corticotropin-like intermediary peptide (CLIP); Lipotropin beta (Beta- DE LPH); Lipotropin gamma (Gamma-LPH); Melanotropin beta (Beta-MSH); DE Beta-endorphin; Met-enkephalin].")
56
II.3.a SWISS-PROT / TREMBL
DE Corticotropin-lipotropin precursor (Pro-opiomelanocortin) (POMC) DE [Contains: NPP; Melanotropin gamma (Gamma-MSH); Corticotropin DE (Adrenocorticotropic hormone) (ACTH); Melanotropin alpha (Alpha-MSH); DE Corticotropin-like intermediary peptide (CLIP); Lipotropin beta (Beta- DE LPH); Lipotropin gamma (Gamma-LPH); Melanotropin beta (Beta-MSH); DE Beta-endorphin; Met-enkephalin]. Liste des peptides produits après clivage de la protéine initiale
 (POMC) DE [Contains: NPP; Melanotropin gamma (Gamma-MSH); Corticotropin. DE (Adrenocorticotropic hormone) (ACTH); Melanotropin alpha (Alpha-MSH); DE Corticotropin-like intermediary peptide (CLIP); Lipotropin beta (Beta- DE LPH); Lipotropin gamma (Gamma-LPH); Melanotropin beta (Beta-MSH); DE Beta-endorphin; Met-enkephalin]. Liste des peptides produits après clivage de la protéine initiale.")
57
II.3.a SWISS-PROT / TREMBL
ID HXK1_ARATH STANDARD; PRT; AA. AC Q42525; Q42535; DT 01-NOV-1997 (Rel. 35, Created) DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 16-OCT-2001 (Rel. 40, Last annotation update) DE Hexokinase 1 (EC ). GN HXK1 OR AT4G29130 OR F19B OS Arabidopsis thaliana (Mouse-ear cress). OC Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; OC Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; OC eurosids II; Brassicales; Brassicaceae; Arabidopsis. OX NCBI_TaxID=3702; gènes
 DT 16-OCT-2001 (Rel. 40, Last sequence update) DT 16-OCT-2001 (Rel. 40, Last annotation update) DE Hexokinase 1 (EC ). GN HXK1 OR AT4G29130 OR F19B OS Arabidopsis thaliana (Mouse-ear cress). OC Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; OC Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots; Rosidae; OC eurosids II; Brassicales; Brassicaceae; Arabidopsis. OX NCBI_TaxID=3702; gènes.")
58
II.3.a SWISS-PROT / TREMBL
Travaux pratiqués sur la séquence par les auteurs. Ici, le commentaire indique que seule la séquence nucléique est expérimentale par conséquent la séquence protéique est conceptuelle RN [1] RP SEQUENCE FROM N.A. RC STRAIN=CV. LANDSBERG ERECTA; RX MEDLINE= ; PubMed= ; RA Dai N., Schaffer A.A., Petreikov M., Granot D.; RT "Arabidopsis thaliana hexokinase cDNA isolated by complementation of RT yeast cells."; RL Plant Physiol. 108: (1995).
.")
59
II.3.a SWISS-PROT / TREMBL
RN [1] RP SEQUENCE FROM N.A. RC STRAIN=CV. LANDSBERG ERECTA; RX MEDLINE= ; PubMed= ; RA Dai N., Schaffer A.A., Petreikov M., Granot D.; RT "Arabidopsis thaliana hexokinase cDNA isolated by complementation of RT yeast cells."; RL Plant Physiol. 108: (1995). Reference comment : origine biologique de la séquence (organisme, plasmide, tissu)
. Reference comment : origine biologique. de la séquence (organisme, plasmide, tissu)")
60
II.3.a SWISS-PROT / TREMBL
Blocs de commentaires CC -!- CATALYTIC ACTIVITY: ATP + D-hexose = ADP + D-hexose 6-phosphate. CC -!- SUBCELLULAR LOCATION: CHLOROPLAST OUTER ENVELOPE; CYTOPLASMIC SIDE (BY SIMILARITY). CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI GAALLAASHS LYLEDS //
. CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM 4 24 POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE 496 AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD. TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM. SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK. LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI. GAALLAASHS LYLEDS. //")
61
Topic Description ALTERNATIVE PRODUCTS Description of the existence of related protein sequence(s) produced by alternative splicing of the same gene or by the use of alternative initiation codons BIOTECHNOLOGY Description of the use of a specific protein in a biotechnological process CATALYTIC ACTIVITY Description of the reaction(s) catalyzed by an enzyme [1] CAUTION This topic warns you about possible errors and/or grounds for confusion COFACTOR Description of an enzyme cofactor DATABASE Description of a cross-reference to a network database/resource for a specific protein [2] DEVELOPMENTAL STAGE Description of the developmental specific expression of a protein DISEASE Description of the disease(s) associated with a deficiency of a protein DOMAIN Description of the domain structure of a protein ENZYME REGULATION Description of an enzyme regulatory mechanism FUNCTION General description of the function(s) of a protein INDUCTION Description of the compound(s) which stimulate the synthesis of a protein MASS SPECTROMETRY Reports the exact molecular weight of a protein or part of a protein as determined by mass spectrometric methods [3] MISCELLANEOUS Any comment which does not belong to any of the other defined topics PATHWAY Description of the metabolic pathway(s) with which a protein is associated PHARMACEUTICAL Description of the use of a specific protein as a pharmaceutical drug POLYMORPHISM Description of polymorphism(s) PTM Description of a posttranslational modification SIMILARITY Description of the similaritie(s) (sequence or structural) of a protein with other proteins SUBCELLULAR LOCATION Description of the subcellular location of the mature protein SUBUNIT Description of the quaternary structure of a protein TISSUE SPECIFICITY Description of the tissue specificity of a protein
 produced by alternative splicing of the same gene or by the use of alternative initiation codons. BIOTECHNOLOGY Description of the use of a specific protein in a biotechnological process. CATALYTIC ACTIVITY Description of the reaction(s) catalyzed by an enzyme [1] CAUTION This topic warns you about possible errors and/or grounds for confusion. COFACTOR Description of an enzyme cofactor. DATABASE Description of a cross-reference to a network database/resource for a specific protein [2] DEVELOPMENTAL STAGE Description of the developmental specific expression of a protein. DISEASE Description of the disease(s) associated with a deficiency of a protein. DOMAIN Description of the domain structure of a protein. ENZYME REGULATION Description of an enzyme regulatory mechanism. FUNCTION General description of the function(s) of a protein. INDUCTION Description of the compound(s) which stimulate the synthesis of a protein. MASS SPECTROMETRY Reports the exact molecular weight of a protein or part of a protein as determined by mass spectrometric methods [3] MISCELLANEOUS Any comment which does not belong to any of the other defined topics. PATHWAY Description of the metabolic pathway(s) with which a protein is associated. PHARMACEUTICAL Description of the use of a specific protein as a pharmaceutical drug. POLYMORPHISM Description of polymorphism(s) PTM Description of a posttranslational modification. SIMILARITY Description of the similaritie(s) (sequence or structural) of a protein with other proteins. SUBCELLULAR LOCATION Description of the subcellular location of the mature protein. SUBUNIT Description of the quaternary structure of a protein. TISSUE SPECIFICITY Description of the tissue specificity of a protein.")
62
II.3.a SWISS-PROT / TREMBL
CC -!- CATALYTIC ACTIVITY: ATP + D-hexose = ADP + D-hexose 6-phosphate. CC -!- SUBCELLULAR LOCATION: CHLOROPLAST OUTER ENVELOPE; CYTOPLASMIC SIDE (BY SIMILARITY). CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI GAALLAASHS LYLEDS // Data base cross-reference
. CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM 4 24 POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE 496 AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD. TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM. SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK. LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI. GAALLAASHS LYLEDS. // Data base cross-reference.")
63
II.3.a SWISS-PROT / TREMBL
CC -!- CATALYTIC ACTIVITY: ATP + D-hexose = ADP + D-hexose 6-phosphate. CC -!- SUBCELLULAR LOCATION: CHLOROPLAST OUTER ENVELOPE; CYTOPLASMIC SIDE (BY SIMILARITY). CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI GAALLAASHS LYLEDS //
. CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM 4 24 POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE 496 AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD. TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM. SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK. LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI. GAALLAASHS LYLEDS. //")
64
II.3.a SWISS-PROT / TREMBL
CC -!- CATALYTIC ACTIVITY: ATP + D-hexose = ADP + D-hexose 6-phosphate. CC -!- SUBCELLULAR LOCATION: CHLOROPLAST OUTER ENVELOPE; CYTOPLASMIC SIDE (BY SIMILARITY). CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI GAALLAASHS LYLEDS //
. CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM 4 24 POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE 496 AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD. TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM. SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK. LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI. GAALLAASHS LYLEDS. //")
65
II.3.a SWISS-PROT / TREMBL
CC -!- CATALYTIC ACTIVITY: ATP + D-hexose = ADP + D-hexose 6-phosphate. CC -!- SUBCELLULAR LOCATION: CHLOROPLAST OUTER ENVELOPE; CYTOPLASMIC SIDE (BY SIMILARITY). CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI GAALLAASHS LYLEDS //
. CC -!- SIMILARITY: BELONGS TO THE HEXOKINASE FAMILY. DR EMBL; U28214; AAB ; -. DR InterPro; IPR001312; Hexokinase. DR Pfam; PF00349; hexokinase; 1. DR PROSITE; PS00378; HEXOKINASES; 1. KW Transferase; Kinase; Glycolysis; ATP-binding; Transmembrane; Chloroplast. FT TRANSMEM 4 24 POTENTIAL. FT NP_BIND ATP (POTENTIAL). FT DOMAIN GLUCOSE-BINDING (POTENTIAL). SQ SEQUENCE 496 AA; MW; 6DC81CE114E0B52B CRC64; MGKVAVGATV VCTAAVCAVA VLVVRRRMQS SGKWGRVLAI LKAFEEDCAT PISKLRQVAD. TLDFESLNPG EQILEKIISG MYLGEILRRV/…../ LLKMAEDAAF FGDTVPSKLR IPFIIRTPHM. SAMHNDTSPD LKIVGSKIKD ILEVPTTSLK MRKVVISLCN IIATRGARLS AAGIYGILKK. LGRDTTKDEE VQKSVIAMDG GLFEHYTQFS ECMESSLKEL LGDEASGSVE VTHSNDGSGI. GAALLAASHS LYLEDS. //")
66
II.3.b Annotations des séquences de Swiss-Prot
Elles concernent les points suivants : Fonction(s) de la protéine Modifications post-traductionnelles (acétylation, phosphorylation,…) Domaines et sites (liaison au calcium, à l’ATP, doigts de zinc, …) Structure secondaire Structure quaternaire (homodimère, hétérotrimère, …) Similitudes avec d’autres protéines Maladies associées à une protéine Conflits sur la séquence, existence de variants, … Sources de l’information Articles concernant une nouvelle séquence Article de synthèse sur les familles de protéines Groupe d’experts Les mises à jour sont régulières La redondance est limitée au mieux
 de la protéine. Modifications post-traductionnelles (acétylation, phosphorylation,…) Domaines et sites (liaison au calcium, à l’ATP, doigts de zinc, …) Structure secondaire. Structure quaternaire (homodimère, hétérotrimère, …) Similitudes avec d’autres protéines. Maladies associées à une protéine. Conflits sur la séquence, existence de variants, … Sources de l’information. Articles concernant une nouvelle séquence. Article de synthèse sur les familles de protéines. Groupe d’experts. Les mises à jour sont régulières. La redondance est limitée au mieux.")
67
II.3.c Quelques statistiques sur Swiss-Prot

68
II.3.c Quelques statistiques sur Swiss-Prot

69
II.3.c Quelques statistiques sur Swiss-Prot

70
II.3.c Quelques statistiques sur Swiss-Prot

71
II Les banques de données
II.4 Les banques d’alignements et de motifs

72
II.4 Les banques d’alignements et de motifs
Définitions Domaine : portion d'une protéine supposée avoir un repliement indépendant du reste de la protéine, et posséder une fonction spécifique. Motif : segment court et conservé d'une séquence nucléique ou protéique. Les motifs sont fréquemment des parties hautement conservées des domaines. Tout commence par des alignements multiples Alignement : Processus par lequel deux séquences sont comparées afin d'obtenir le plus de correspondances (identités ou substitutions ) possibles entre les nucléotides ou acides aminés qui les composent. Alignement global : alignement des deux séquences sur toute leur longueur. (Gap) Alignement local : alignement des deux séquences sur une portion de leur longueur. (Fasta et Blast) Alignement optimal : alignement de deux séquences de façon à obtenir le plus haut score possible. (Needleman et Wunsch) Alignement multiple : alignement global de trois ou plus de trois séquences. (ClustalW)
 possibles entre les nucléotides ou acides aminés qui les composent. Alignement global : alignement des deux séquences sur toute leur longueur. (Gap) Alignement local : alignement des deux séquences sur une portion de leur longueur. (Fasta et Blast) Alignement optimal : alignement de deux séquences de façon à obtenir le plus haut score possible. (Needleman et Wunsch) Alignement multiple : alignement global de trois ou plus de trois séquences. (ClustalW)")
73
II.4 Les banques d’alignements et de motifs
Il existe de nombreuses banques d’alignements et de motifs PROSITE : SIB, Dictionnaire de sites et motifs protéiques (expressions régulières) Profiles : ISREC, Lausanne, matrices pondérées (profils) PRINTS : UCL London, (Protein Motif Fingerprint Database). Une empreinte (fingerprint) est un groupe conservé de motifs utilisé pour caractériser une famille de protéines Pfam : Sanger centre, Collection de familles alignées de protéines, générées automatiquement ou semi-automatiquement par la méthode "Hidden Markov Models" (HMMs). BLOCKS : FHCRC Seattle, « blocks », alignements multiples de segments sans insertions, correspondant aux régions les mieux conservées de Prosite ProDom : (PROtein DOMain Database) INRA, Toulouse, compilation automatisée des domaines homologues (alignements multiples et consensus) détectés dans Swiss-prot
 Profiles : ISREC, Lausanne, matrices pondérées (profils) PRINTS : UCL London, (Protein Motif Fingerprint Database). Une empreinte (fingerprint) est un groupe conservé de motifs utilisé pour caractériser une famille de protéines. Pfam : Sanger centre, Collection de familles alignées de protéines, générées automatiquement ou semi-automatiquement par la méthode Hidden Markov Models (HMMs). BLOCKS : FHCRC Seattle, « blocks », alignements multiples de segments sans insertions, correspondant aux régions les mieux conservées de Prosite. ProDom : (PROtein DOMain Database) INRA, Toulouse, compilation automatisée des domaines homologues (alignements multiples et consensus) détectés dans Swiss-prot.")
74
II. 4 Les banques d’alignements et de motifs II. 4
II.4 Les banques d’alignements et de motifs II.4.a PROSITE / PROFILES: SIB, expressions régulières Alignement multiple de séquences homologues issues de Swiss-Prot Détermination manuelle d’une expression consensus Affinage du consensus contre Swiss-Prot : Le consensus doit permettre de récupérer les séquences qui ont servi à le construire. Il y a des faux positifs ainsi que des faux négatifs. « pattern / profile » C-x(3)-[LIVMFY]-x(5)-[LIVMFY]-x(3)-[DENQ]-[LIVMFY]-x(10)- C-x(3)-C-T-x(4)-C-x-[LIVMFY]-F-x-[FY]-x(13,14)-C-x- [LIVMFY]-[RK]-x-[ST]-x(14,15)-S-G-x-[ST]-[LIVMFY]-x(2)-C Version 16.53, of 06-Dec-2001 (contient 1104 fiches documentation décrivant 1494 « patterns », règles et profils/matrices).
-[LIVMFY]-x(5)-[LIVMFY]-x(3)-[DENQ]-[LIVMFY]-x(10)- C-x(3)-C-T-x(4)-C-x-[LIVMFY]-F-x-[FY]-x(13,14)-C-x- [LIVMFY]-[RK]-x-[ST]-x(14,15)-S-G-x-[ST]-[LIVMFY]-x(2)-C. Version 16.53, of 06-Dec-2001 (contient 1104 fiches documentation décrivant 1494 « patterns », règles et profils/matrices).")
76
II. 4 Les banques d’alignements et de motifs II. 4
II.4 Les banques d’alignements et de motifs II.4.b PRINTS : UCL London, empreintes (fingerprints) Alignement local Identification manuelle d’un « Fingerprint » : ensemble de 1 à n motifs Affinage contre OWL « Fingerprint » composé d’un jeu de motifs Version 32.0 de PRINTS contient 1600 entrées, codant 9800 motifs.
 Alignement local. Identification manuelle d’un. « Fingerprint » : ensemble de 1 à n motifs. Affinage contre OWL. « Fingerprint » composé d’un jeu de motifs. Version 32.0 de PRINTS contient 1600 entrées, codant 9800 motifs.")
77
II. 4 Les banques d’alignements et de motifs II. 4
II.4 Les banques d’alignements et de motifs II.4.c Pfam : Sanger centre, HMMs profiles Alignement multiple édité manuellement Un profil « HMM » en est dérivé Progression aléatoire estimant la probabilité de transition à chaque étape et utilisant la technique d'apprentissage Alignement pleine longueur final Pfam A : Alignements précis vérifiés, annotés (3071 familles, séquences) Pfam B : Clustering automatique de Swiss Prot / Trembl, non annoté (57477, )
 Pfam B : Clustering automatique de Swiss Prot / Trembl, non annoté (57477, )")
78
Transition 1 -> 2 Prob(C ->G) 2 0.4 Prob(C->C) 2 0.4 Prob(G->C) 1 0.2 Prob(G->G) 0 0
 Prob(C->C) Prob(G->C) Prob(G->G) 0 0")
79
Modèle de Markov début fin début fin
émission début transition fin 0.4 C 0.8 C 0.6 0.8 début 0.4 fin 0.2 G 0.2 G 0.4 0.2 P(CG) = 0.8 * 0.8 * 0.4 * 0.4 = 0.102 P(GC) = 0.2 * 0.2 * 0.2 * 0.6 = 0.004 P(GA) = 0.2 * 0.2 * 0 * 0 = 0
 = 0.8 * 0.8 * 0.4 * 0.4 = P(GC) = 0.2 * 0.2 * 0.2 * 0.6 = P(GA) = 0.2 * 0.2 * 0 * 0 = 0.")
80
II. 4 Les banques d’alignements et de motifs II. 4
II.4 Les banques d’alignements et de motifs II.4.c Pfam : Sanger centre, HMMs profiles Pfam entry: Glyco_hydro_68 Accession number: PF02435 Definition: Levansucrase/Invertase Author: Mian N, Bateman A Alignment method of seed: Clustalw Source of seed members: Pfam-B_2011 (release 5.4) Gathering cutoffs: Trusted cutoffs: Noise cutoffs: HMM build command line: hmmbuild -F HMM SEED HMM build command line: hmmcalibrate --seed 0 HMM Reference Number: [1] Reference Medline: Reference Title: Cloning, nucleotide sequence, and expression in Escherichia Reference Title: coli of levansucrase genes from the plant pathogens Reference Title: Pseudomonas syringae pv. glycinea and P. syringae pv. Reference Title: phaseolicola. Reference Author: Hettwer U, Jaeckel FR, Boch J, Meyer M, Rudolph K, Ullrich Reference Author: MS; Reference Location: Appl Environ Microbiol 1998;64: Database Reference INTERPRO; IPR003469; Comment: This Pfam family consists of the glycosyl hydrolase 68 family, Comment: including several bacterial levansucrase enzymes, and invertase from Comment: zymomonas. Number of members: 14
 Gathering cutoffs: Trusted cutoffs: Noise cutoffs: HMM build command line: hmmbuild -F HMM SEED. HMM build command line: hmmcalibrate --seed 0 HMM. Reference Number: [1] Reference Medline: Reference Title: Cloning, nucleotide sequence, and expression in Escherichia. Reference Title: coli of levansucrase genes from the plant pathogens. Reference Title: Pseudomonas syringae pv. glycinea and P. syringae pv. Reference Title: phaseolicola. Reference Author: Hettwer U, Jaeckel FR, Boch J, Meyer M, Rudolph K, Ullrich. Reference Author: MS; Reference Location: Appl Environ Microbiol 1998;64: Database Reference INTERPRO; IPR003469; Comment: This Pfam family consists of the glycosyl hydrolase 68 family, Comment: including several bacterial levansucrase enzymes, and invertase from. Comment: zymomonas. Number of members: 14.")
81
II.4 Les banques d’alignements et de motifs II.4.d PRODOM
Swiss Prot + Trembl PSI-BLAST récursifs Domaines, consensus Proteines utilisées pour ProDom : (SwissProt /TREMBL – Mai 2001) domaines avec au moins 2 sequences domaines 339763 101957 283772
 domaines avec au moins 2 sequences. domaines")
82
II. 4 Les banques d’alignements et de motifs II. 4
II.4 Les banques d’alignements et de motifs II.4.e BLOCKS : FHCRC Seattle, « blocks » Prosite (4034) ProDom (1066) Pfam(2258) Domo(306) Groupes de protéines Alignement local sans gap avec un germe de trois acides aminés BLOCKS Calibration contre Swiss Prot Version 13.0 (08/001): 8656 blocks représentant 2101 groupes
 ProDom (1066) Pfam(2258) Domo(306) Groupes de protéines. Alignement local sans gap avec un germe de trois acides aminés. BLOCKS. Calibration contre Swiss Prot. Version 13.0 (08/001): 8656 blocks représentant 2101 groupes.")
83
II. 4 Les banques d’alignements et de motifs II. 4
II.4 Les banques d’alignements et de motifs II.4.f Quelle banque pour quel résultat ? Recherche des membres de super-familles très divergentes Profiles, Pfam Recherche des membres de sous-familles Prints Recherche de motifs courts Prosite, Blocks

84
II. 4 Les banques d’alignements et de motifs II. 4
II.4 Les banques d’alignements et de motifs II.4.f INTERPRO la base intégrative INTERPRO : Pfam, Prints, Prosite, Swiss Prot / Trembl sont intégrées dans une hiérarchie formant des familles.

85
II.5 La recherche dans les banques
Mots-clés et critères Numéro d’accession Nom de séquence Organisme Définition Taille d’une séquence Séquence Recherche de séquences homologues Recherche de domaines ou de motifs

86
II.5 La recherche dans les banques II.5.a ENTREZ

88
II. 5 La recherche dans les banques II. 5
II.5 La recherche dans les banques II.5.b SRS (Sequence Retrieval System)
")
93
Recherche de similarités entre séquences biologiques
Objectifs Recherche d’informations sur la fonction biologique Etude de la structure (motifs, domaines, …) Informations sur l’évolution des séquences (phylogénie) Constitue en générale la première étape de l’étude d’une séquence nouvelle
 Informations sur l’évolution des séquences (phylogénie) Constitue en générale la première étape. de l’étude d’une séquence nouvelle.")
94
Deux gènes sont homologues s’ils ont un gène ancêtre en commun
Recherche de similarités entre séquences biologiques La recherche de similarités permet de mettre en évidence les régions proches de deux séquences Similarité et homologie : Deux gènes sont homologues s’ils ont un gène ancêtre en commun On doit donc parler de recherche de similarités. Si une protéine partage 25 % d’identité sur une longueur de 100 acides aminés avec une autre, on pourra parler d’homologie

95
Recherche de similarités entre séquences biologiques
On peut comparer des séquences nucléiques ou des séquences protéiques La probabilité est plus forte de trouver de la similarité par hasard dans les comparaisons ADN/ADN car on se restreint à 4 nucléotides contre 20 acides aminés Séquence de 10 bases -> 410 séquences possibles soit Les banques publiques contiennent nucléotides à partir desquels on peut extraire environ séquences de 10 bases. Une séquence particulière de 10 bases peut donc s’y trouver 6600 fois simplement par hasard. Ainsi obtenir 100 % d’identité sur 10 bases peut ne pas avoir beaucoup de signification biologique et n’être que le fruit du hasard Séquence de 10 aa -> 2010 séquences possibles soit 10,

96
Recherche de similarités entre séquences biologiques
La recherche de similarités repose sur des processus évolutifs : les mutations Séq. 1 G T C A G substitution Séq. 2 G T T A G suppression Séq. 3 G T A G insertion Séq. 4 G T A T G

97
Recherche de similarités entre séquences biologiques
La mise en évidence de similarités réclame d’aligner des séquences. 3 identités, 1 substitution et 2 indels (INsertion-DELétion) 4 identités et 2 indels Séq. 1 G T C A _ G Séq. 4 G _ T A T G Séq. 1 G T C A _ G Séq. 4 G T _ A T G
 4 identités et 2 indels. Séq. 1 G T C A _ G. Séq. 4 G _ T A T G. Séq. 1 G T C A _ G. Séq. 4 G T _ A T G.")
98
Recherche de similarités entre séquences biologiques
Comment évaluer un alignement : le score G T C A G G T A T G Appariement = 1, 0 sinon Score = S(i) = 3 1 1 –1/4 1 –1/4 1 G T C A G G T A T G Appariement = 1; INDEL = -0,25; 0 sinon Score = S(i) - P(i) = 3,5
 = –1/4 1 –1/4 1. G T C A G. G T A T G. Appariement = 1; INDEL = -0,25; 0 sinon. Score = S(i) - P(i) = 3,5.")
99
Recherche de similarités entre séquences biologiques
CGCCGGTGTACTGCA-C-TGGCGTG--TCA CGCCGG-G-ACCGCAGCATGGCGGGCATCA Cet alignement nécessite deux insertions (GAP) consécutives. Deux paramètres décrivent un gap sa création sa longueur
 consécutives. Deux paramètres décrivent un gap. sa création. sa longueur.")
100
Recherche de similarités entre séquences biologiques
Il est possible d’imposer une pénalité résumant les deux paramètres P = A + B * L P pénalité A pénalité d’ouverture de gap B pénalité d’extension de gap L longueur du gap Si on prend A grand et B petit on favorise des alignements comportant peu de gaps mais de taille importante. Si on prend A petit et B grand alors les alignements avec plusieurs gaps mais de courte longueur auront de meilleurs scores que ceux de la situation précédente (moins en accord avec la réalité biologique)
")
101
Recherche de similarités entre séquences biologiques
Les matrices de substitution Pour les séquences nucléiques : la matrice unitaire A T G C 1 D’autres matrices peuvent être utilisées basées par exemple sur les fréquences de substitution observées sur un jeu de séquences

102
Recherche de similarités entre séquences biologiques
Pour les acides aminés on peut également imaginer une matrice unitaire. Mais une telle matrice serait trop sélective. En effet, remplacer une leucine par une valine doit avoir moins d’impact sur la structure et la fonction d’une protéine que de la remplacer par une proline. Par ailleurs, les séquences protéiques étant soumises à la pression sélective, les mutations que l’on observe entre les séquences d’une même protéine d’organismes différents sont mieux tolérées que les autres. Ainsi, il faut imaginer d’autre matrices pour que le score associé au remplacement d’un acide aminé par un autre tienne compte des conséquences que cette substitution peut avoir sur la fonction ou la structure de la protéine.

103
Recherche de similarités entre séquences biologiques
Les matrices protéiques liées à l’évolution Elles représentent les échanges possibles ou acceptables d’un acide aminé par un autre lors de l’évolution des protéines Les matrices PAM (Point Accepted Mutation) Etude de 71 familles de protéines contenant des séquences (1300) très semblables, donc s’alignant facilement A partir des alignements, on calcule une matrice de probabilité ou chaque élément désigne la probabilité qu’un acide aminé A soit remplacé par un acide aminé B durant une étape d’évolution Cette matrice correspond à un temps d’évolution autorisant 1 mutation pour 100 sites : on parle de 1PAM ou PAM-1 (après un reformatage). Si l’on multiplie cette matrice par elle même X fois, on obtient une matrice PAM-X correspondant à un temps d’évolution plus long. La matrice PAM-250 semble la plus adaptée pour distinguer des protéines proches de celles similaires par hasard. Inconvénient : tous les sites sont considérés comme équiprobables vis à vis du taux de mutation => faux
 Etude de 71 familles de protéines contenant des séquences (1300) très semblables, donc s’alignant facilement. A partir des alignements, on calcule une matrice de probabilité ou chaque élément désigne la probabilité qu’un acide aminé A soit remplacé par un acide aminé B durant une étape d’évolution. Cette matrice correspond à un temps d’évolution autorisant 1 mutation pour 100 sites : on parle de 1PAM ou PAM-1 (après un reformatage). Si l’on multiplie cette matrice par elle même X fois, on obtient une matrice PAM-X correspondant à un temps d’évolution plus long. La matrice PAM-250 semble la plus adaptée pour distinguer des protéines proches de celles similaires par hasard. Inconvénient : tous les sites sont considérés comme équiprobables vis à vis du taux de mutation => faux.")
104
Recherche de similarités entre séquences biologiques
Les matrices BLOSUM (BLOcks Substitution Matrix) Les matrices PAM sont construites à partir d’alignements globaux de protéines très semblables. Les BLOSUM sont élaborées à partir de BLOCKS (séquence issues d’alignements multiples sans insertion délétion de courtes régions conservées) Ces blocs permettent de rassembler toutes les séquences ayant un taux d’identité minimum au sein de leur bloc. On en déduit, pour le taux d’identité en question, une matrice de probabilité de substitution d’un acide aminé par un autre. A chaque taux d’identité correspond une matrice BLOSUM particulière. BLOSUM60 : 60 % d’identité Non basées sur un modèle évolutif (bien qu’implicite) Donnent de meilleurs résultats que PAM Construites à partir d’un plus grand nombre de séquences Basées uniquement sur les régions les plus homologues (blocks)
 Les matrices PAM sont construites à partir d’alignements globaux de protéines très semblables. Les BLOSUM sont élaborées à partir de BLOCKS (séquence issues d’alignements multiples sans insertion délétion de courtes régions conservées) Ces blocs permettent de rassembler toutes les séquences ayant un taux d’identité minimum au sein de leur bloc. On en déduit, pour le taux d’identité en question, une matrice de probabilité de substitution d’un acide aminé par un autre. A chaque taux d’identité correspond une matrice BLOSUM particulière. BLOSUM60 : 60 % d’identité. Non basées sur un modèle évolutif (bien qu’implicite) Donnent de meilleurs résultats que PAM. Construites à partir d’un plus grand nombre de séquences. Basées uniquement sur les régions les plus homologues (blocks)")
105
Recherche de similarités entre séquences biologiques
Les matrices liées aux propriétés physico-chimiques Matrice basée sur des mesures d’énergie libre de transfert de l’eau à l’éthanol des acides aminés (Levitt, 1976) Matrice de structure secondaire basée sur la propension d’un acide aminé à se trouver dans une hélice, un feuillet ou un coude (Levin, 1986) Matrice basée sur les structure 3D : permet de comparer des protéines assez éloignées ( la structure 3D est plus conservée que la structure primaire => deux protéines peuvent partager la même structure 3D et donc posséder des fonctions biologiques analogues tout en ayant des séquences très différentes => convergence)
 Matrice de structure secondaire basée sur la propension d’un acide aminé à se trouver dans une hélice, un feuillet ou un coude (Levin, 1986) Matrice basée sur les structure 3D : permet de comparer des protéines assez éloignées ( la structure 3D est plus conservée que la structure primaire => deux protéines peuvent partager la même structure 3D et donc posséder des fonctions biologiques analogues tout en ayant des séquences très différentes => convergence)")
106
Recherche de similarités entre séquences biologiques
Le choix d’une matrice BLOSUM élevées (80) et PAM faibles (1) permettent de comparer des séquences proches et courtes BLOSUM faibles (45) et PAM élevées (250) pour les séquences plus divergentes et plus longues Pour démarrer une étude il faut utiliser la BLOSUM 62 ou la PAM 120
 et PAM faibles (1) permettent de comparer des séquences proches et courtes. BLOSUM faibles (45) et PAM élevées (250) pour les séquences plus divergentes et plus longues. Pour démarrer une étude il faut utiliser la BLOSUM 62 ou la PAM 120.")
107
Recherche de similarités entre séquences biologiques
Les outils d’alignement Le « dot plot » A T G C X

108
Recherche de similarités entre séquences biologiques
Les outils d’alignement Le « dot plot » A T G C X

109
Recherche de similarités entre séquences biologiques
Les outils d’alignement Le « dot plot » A T G C X

110
Recherche de similarités entre séquences biologiques
Les outils d’alignement Le « dot plot » A T G C X

111
Recherche de similarités entre séquences biologiques
Les outils d’alignement Le « dot plot » A T G C X

112
Recherche de similarités entre séquences biologiques
Les outils d’alignement Le « dot plot » A T G C X

113
Recherche de similarités entre séquences biologiques
L’alignement optimal Exemple : deux séquences à comparer ATGTAATGCATA TATGTGAAT Scores identité +1 gap -1 extension -1 Alignement optimal par glissement score = 5 A T G T A A T G C A T G T A T G T G A A T Alignement optimal avec insertion score = 6 A T G T - A A T G C A T G T A T G T G A A T

114
Recherche de similarités entre séquences biologiques
L’alignement optimal Algorithme de Needleman et Wunsch Alignement optimal global de deux séquences Algorithme de Smith et Waterman Alignement optimal local de deux séquences Ces algorithmes sont les meilleurs mais ils sont très coûteux en temps de calcul. Ils ne sont donc pas utilisés pour la recherche de similarités entre une séquence et une banque de séquences

115
Recherche de similarités entre séquences biologiques
La recherche de similarités dans les banques FASTA Identification rapide de zone d’identité entre la séquence requête et les séquences banque. Bonne sensibilité car il prend en compte les INDELs Les « hits » ou résultats sont fournis avec un Z-score et une E-value Z-score = (s-m)/e S : score observé M : moyenne des scores aléatoires E : écart type des scores aléatoires E-value Plus elle est faible et moins on a de chance d’avoir trouver par hasard l’alignement observé E-value < 0,01 : séquences homologues E-value : séquences plus lointaines
/e. S : score observé. M : moyenne des scores aléatoires. E : écart type des scores aléatoires. E-value. Plus elle est faible et moins on a de chance d’avoir trouver par hasard l’alignement observé. E-value < 0,01 : séquences homologues. E-value 1-10 : séquences plus lointaines.")
116
Recherche de similarités entre séquences biologiques
La recherche de similarités dans les banques BLAST (Basic Local Alignment Search Tool) Développé au NCBI Basé sur un modèle statistique L’unité de base de l’algorithme est le HSP (High-scoring Segment Pair) Il s’agit d’une région de similitude la plus longue possible entre deux séquences ayant un score supérieur ou égal à un score seuil. Il peut y avoir plusieurs HSP issus de la comparaison de deux séquences. Un deuxième score MSP (Maximal-scoring Segment Pair) est le meilleur score obtenu parmi tous les couples possibles que peuvent produire deux séquences. Les méthodes statistiques sont utilisées pour évaluer la qualité des HSPs et MSPs.
 Développé au NCBI. Basé sur un modèle statistique. L’unité de base de l’algorithme est le HSP (High-scoring Segment Pair) Il s’agit d’une région de similitude la plus longue possible entre deux séquences ayant un score supérieur ou égal à un score seuil. Il peut y avoir plusieurs HSP issus de la comparaison de deux séquences. Un deuxième score MSP (Maximal-scoring Segment Pair) est le meilleur score obtenu parmi tous les couples possibles que peuvent produire deux séquences. Les méthodes statistiques sont utilisées pour évaluer la qualité des HSPs et MSPs.")
117
Recherche de similarités entre séquences biologiques
La recherche de similarités dans les banques BLAST (Basic Local Alignment Search Tool) Il existe cinq programmes BLASTN (séquence nucléique contre banque nucléique) BLASTP (séquence protéique contre banque protéique) BLASTX (séquence nucléique traduite en 6 phases contre banque protéique) TBLASTN (séquence protéique contre banque nucléique traduite en 6 phases) TBLASTX (séquence nucléique traduite dans les 6 phases contre banque traduite dans les 6 phases) Les étapes de l’algorithme Faire une liste de tous les mots de longueur X dans la séquence Par défaut X = 3 pour les protéines et 11 pour les acides nucléiques, l’utilisateur peut modifier ces paramètres Comparer ces mots avec les séquences de la banques pour identifier les séquences identiques (les « hits »)
 Il existe cinq programmes. BLASTN (séquence nucléique contre banque nucléique) BLASTP (séquence protéique contre banque protéique) BLASTX (séquence nucléique traduite en 6 phases contre banque protéique) TBLASTN (séquence protéique contre banque nucléique traduite en 6 phases) TBLASTX (séquence nucléique traduite dans les 6 phases contre banque traduite dans les 6 phases) Les étapes de l’algorithme. Faire une liste de tous les mots de longueur X dans la séquence. Par défaut X = 3 pour les protéines et 11 pour les acides nucléiques, l’utilisateur peut modifier ces paramètres. Comparer ces mots avec les séquences de la banques pour identifier les séquences identiques (les « hits »)")
118
Recherche de similarités entre séquences biologiques
La recherche de similarités dans les banques BLAST (Basic Local Alignment Search Tool) Extension du segment identique lorsque cela est possible, dans les deux directions de manière à ce que le score cumulé puisse être amélioré. L’extension est stoppée dans trois cas : Si le score cumulé descend d’une quantité x donnée par rapport à la valeur maximale qu’il avait atteint Si le score cumulé devient inférieur ou égal à zéro Si la fin de l’une des deux séquences est atteinte
 Extension du segment identique lorsque cela est possible, dans les deux directions de manière à ce que le score cumulé puisse être amélioré. L’extension est stoppée dans trois cas : Si le score cumulé descend d’une quantité x donnée par rapport à la valeur maximale qu’il avait atteint. Si le score cumulé devient inférieur ou égal à zéro. Si la fin de l’une des deux séquences est atteinte.")
119
Recherche de similarités entre séquences biologiques
Séquence requête (query) Liste des mots de longueur l Comparaison des mots avec les séquences de la banque (subject) et identification des « hits » exacts Extension des HSPs
 Liste des mots de longueur l. Comparaison des mots avec les. séquences de la banque (subject) et identification des « hits » exacts. Extension des HSPs.")
120
Recherche de similarités entre séquences biologiques
La recherche de similarités dans les banques BLAST (Basic Local Alignment Search Tool) L’évaluation du « hit » Le score S (bits) : Il est dérivé du score brut de l’alignement. Il a été normalisé dans le but de pouvoir comparer des scores issus de recherches différentes. La E-value (Expected) : nombre d’alignements différents que l’on peut espérer trouver dans la banque avec un score supérieur ou égal à S (probabilité d’observer au hasard ce score). Plus la E-value est faible et plus l’alignement est significatif. La E-value tient compte de la taille de la séquence requête la taille de la banque la composition de la séquence la matrice de substitution utilisée E-value < e-100 => même gène ou allèles ou espèces voisines E-value e-100 e-80 => gènes très proche
 L’évaluation du « hit » Le score S (bits) : Il est dérivé du score brut de l’alignement. Il a été normalisé dans le but de pouvoir comparer des scores issus de recherches différentes. La E-value (Expected) : nombre d’alignements différents que l’on peut espérer trouver dans la banque avec un score supérieur ou égal à S (probabilité d’observer au hasard ce score). Plus la E-value est faible et plus l’alignement est significatif. La E-value tient compte de. la taille de la séquence requête. la taille de la banque. la composition de la séquence. la matrice de substitution utilisée. E-value < e-100 => même gène ou allèles ou espèces voisines. E-value e-100 e-80 => gènes très proche.")
121
Recherche de similarités entre séquences biologiques
La recherche de similarités dans les banques BLAST (Basic Local Alignment Search Tool) Paramètres Taille du mot : plus on l’élève plus la recherche est spécifique et moins elle est sensible Filtres : permet de masquer les régions de faible complexité (séquences répétées, séquences présentes dans de très nombreuses protéines, logiciels SEG et XNU) Matrices : choix de la matrice de score (BLOSUM, PAM, …) EXPECT : définition du score seuil pour la recherche, seuls les alignements dont le score est inférieur à E seront reportés. Plus E est pris faible et plus les résultats seront fiables. GAPS : choix des pénalités d’ouverture et d’extension de gap ouverture extension BLASTP -11 -1 BLASTN -5 -2
 Paramètres. Taille du mot : plus on l’élève plus la recherche est spécifique et moins elle est sensible. Filtres : permet de masquer les régions de faible complexité (séquences répétées, séquences présentes dans de très nombreuses protéines, logiciels SEG et XNU) Matrices : choix de la matrice de score (BLOSUM, PAM, …) EXPECT : définition du score seuil pour la recherche, seuls les alignements dont le score est inférieur à E seront reportés. Plus E est pris faible et plus les résultats seront fiables. GAPS : choix des pénalités d’ouverture et d’extension de gap. ouverture. extension. BLASTP BLASTN")
122
Recherche de similarités entre séquences biologiques
filtre off on matrice PAM35 ou moins PAM70 BLOSUM80 BLOSUM62 L mot 3 ou 2 3 Gap (ouv, ext) 9, 1 10, 1 11, 1 E-value (seuil) 10000 10-100 10 Paramètres recommandés pour une séquence nucléique (infobiogen)
 9, 1. 10, 1. 11, 1. E-value (seuil) Paramètres recommandés pour une séquence nucléique (infobiogen)")
123
Page d’accueil du serveur BLAST au NCBI

134
Recherche de similarités entre séquences biologiques
L’alignement multiple Détecter des régions conservées dans des familles de séquences. Caractériser de nouvelles familles de protéines. Détecter ou démontrer une homologie entre différentes séquences Trouver des amorces de PCR pour amplifier une famille de gènes ou un membre d’une famille de gène Etablir une phylogénie Aider à la modélisation : les algorithmes de prédiction de structure secondaire exploitent très bien les alignements multiples

135
Recherche de similarités entre séquences biologiques
L’alignement multiple Il s’agit d’un processus qui peut être coûteux en temps de calcul Ce temps dépend de trois paramètres Le volume des données à traiter (taille et nombre de séquences) La puissance de l’ordinateur utilisé La puissance de l’algorithme utilisé Il existe deux classes d’algorithme La première dérive de l’algorithme de Needleman et Wunsch On recherche l'alignement multiple qui maximise la somme des scores de chaque alignement pour chaque paire (pour n séquences, il y a n(n-1)/2 paires). Cependant, la taille du problème, en temps et en place mémoire dans l'ordinateur, est proportionnelle au produit des longueurs des séquences : si les N séquences sont de longueur L, la complexité est en LN. Cette complexité croit de façon exponentielle avec le nombre de séquences, elle est donc utilisable avec un petit nombre de séquences mais ne peut répondre à la plupart des besoins.
 La puissance de l’ordinateur utilisé. La puissance de l’algorithme utilisé. Il existe deux classes d’algorithme. La première dérive de l’algorithme de Needleman et Wunsch. On recherche l alignement multiple qui maximise la somme des scores de chaque alignement pour chaque paire (pour n séquences, il y a n(n-1)/2 paires). Cependant, la taille du problème, en temps et en place mémoire dans l ordinateur, est proportionnelle au produit des longueurs des séquences : si les N séquences sont de longueur L, la complexité est en LN. Cette complexité croit de façon exponentielle avec le nombre de séquences, elle est donc utilisable avec un petit nombre de séquences mais ne peut répondre à la plupart des besoins.")
136
Recherche de similarités entre séquences biologiques
L’alignement multiple La deuxième utilise une méthode heuristique Heuristique : méthode donnant rapidement un bon résultat sans que l’on soit assuré que ce soit le meilleur (BLAST en fait partie) C'est l'approche la plus commune. Cette méthode est rapide et dans la plupart des cas, donne de bons résultats. Elle est utilisée par le programme Clustalw. Clustalw commence par aligner deux à deux les séquences et construit l'arbre des relations évolutives entre les séquences. Les nœuds entre les branches représentent les alignements deux à deux et la racine représente l'alignement complet. Une fois cet arbre construit, le programme prend les deux séquences les plus proches et commence l'alignement multiple (l'alignement des séquences les plus proches est le plus fiable). Puis il progresse vers les séquences plus distantes, et remonte ainsi l'arbre. Ce programme est rapide pour un nombre raisonnable de séquences longues et plus lent si on aligne un grand nombre de séquences courtes.
 C est l approche la plus commune. Cette méthode est rapide et dans la plupart des cas, donne de bons résultats. Elle est utilisée par le programme Clustalw. Clustalw commence par aligner deux à deux les séquences et construit l arbre des relations évolutives entre les séquences. Les nœuds entre les branches représentent les alignements deux à deux et la racine représente l alignement complet. Une fois cet arbre construit, le programme prend les deux séquences les plus proches et commence l alignement multiple (l alignement des séquences les plus proches est le plus fiable). Puis il progresse vers les séquences plus distantes, et remonte ainsi l arbre. Ce programme est rapide pour un nombre raisonnable de séquences longues et plus lent si on aligne un grand nombre de séquences courtes.")
137
Soit 4 séquences s1, s2, s3 et s4 1) Réalisation de l'alignement deux à deux des séquences avec calcul d'un score : on obtient donc une distance pour chaque couple. 2) Construction d'une matrice de distances. s1 s2 s3 s4 d3 d1 d4 d5 d2 d6 S4 S3 / \ S1 / \ / S2 / C1 \ / \ C2 \ / \ / C3 3) Elaboration d'un dendrogramme qui donnera l'ordre de l'alignement multiple On considère que d1 < d2 < d3 < d4 <d5 < d6
 Construction d une matrice de distances. s1. s2. s3. s4. d3. d1. d4. d5. d2. d6. S4 S3 / \ S1 / \ / S2 / C1 \ / \ C2 \ / \ / C3. 3) Elaboration d un dendrogramme qui donnera l ordre. de l alignement multiple On considère que d1 < d2 < d3 < d4 <d5 < d6.")
138
4) Construction successives de consensus permettant de remonter dans l'arbre
a S3 S1 C1 b. S2 S4 C2 c C1 C3 C3

139
****** MULTIPLE ALIGNMENT MENU ******
1. Do complete multiple alignment now (Slow/Accurate) On peut utiliser un ancien dendrogramme pour guider l'alignement initial ou seulement produire ce dendrogramme sans aller jusqu'à l'alignement multiple 2. Produce guide tree file only 3. Do alignment using old guide tree file 4. Toggle Slow/Fast pairwise alignments = SLOW 5. Pairwise alignment parameters contrôle la vitesse et la sensibilité de l'alignement initial 6. Multiple alignment parameters contrôle les gaps dans l'alignement multiple final Dans ce menu, on peut choisir la vitesse avec laquelle on veut que l'alignement se fasse : - Méthode lente mais précise : cette méthode est très lente pour un grand nombre (> 100) de longues séquences (>1000 résidus). - Méthode rapide mais plus approximative. 7. Reset gaps before alignment? = OFF cela permet d'effacer tous les gaps d'un alignement et de le refaire en modifiant certaines options (ON). Si cette option est OFF, les nouveaux gaps seront conservés si un second alignement est réalisé (Parfois, un deuxième passage voire un troisième améliore la qualité de l'alignement). 8. Toggle screen display = ON Les résultats sont envoyés à la fois à l'écran et dans un fichier de sortie.
 On peut utiliser un ancien dendrogramme pour guider l alignement initial ou seulement produire ce dendrogramme sans aller jusqu à l alignement multiple 2. Produce guide tree file only 3. Do alignment using old guide tree file. 4. Toggle Slow/Fast pairwise alignments = SLOW. 5. Pairwise alignment parameters contrôle la vitesse et la sensibilité de l alignement initial 6. Multiple alignment parameters contrôle les gaps dans l alignement multiple final. Dans ce menu, on peut choisir la vitesse avec laquelle on veut que l alignement se fasse : - Méthode lente mais précise : cette méthode est très lente pour un grand nombre (> 100) de longues séquences (>1000 résidus). - Méthode rapide mais plus approximative. 7. Reset gaps before alignment = OFF cela permet d effacer tous les gaps d un alignement et de le refaire en modifiant certaines options (ON). Si cette option est OFF, les nouveaux gaps seront conservés si un second alignement est réalisé (Parfois, un deuxième passage voire un troisième améliore la qualité de l alignement). 8. Toggle screen display = ON Les résultats sont envoyés à la fois à l écran et dans un fichier de sortie.")
140
********* PAIRWISE ALIGNMENT PARAMETERS *********
Slow/Accurate alignments: Si on augmente les pénalités d'ouverture et d'extension des gaps, ceux-ci seront moins fréquents (Gap open Penalty) et plus courts (Gap Extension Penalty). 1. Gap Open Penalty : 2. Gap Extension Penalty :0.10 3. Protein weight matrix :Gonnet series matrice de scores donnant la similarité des acides aminés les par rapport aux autres. On peut choisir entre BLOSUM 30,PAM 35, Gonnet 250, la matrice identité ou une matrice personnelle. 4. DNA weight matrix :IUB matrice de scores pour les acides nucléiques. Fast/Approximate alignments: Si on augmente la taille des k-tuples, la vitesse augmente tandis que si on la diminue, c'est la précision qui augmente. [les maximum sont de 2 pour les protéines et 4 pour les acides nucléiques] 5. Gap penalty :3 cela n'a que peut d'influence sur la rapidité de l'alignement sauf pour des valeurs extrêmes. 6. K-tuple (word) size :1 7. No. of top diagonals :5 nombre de k-tuples qui matchent sur chaque diagonale (dans un dotplot imaginaire) et seuls les meilleurs sont utilisés pour l'alignement. Une diminution de ce paramètre rend l'alignement plus rapide tandis qu'une augmentation améliore la sensibilité. 8. Window size :5 9. Toggle Slow/Fast pairwise alignments = SLOW
 et plus courts (Gap Extension Penalty). 1. Gap Open Penalty : Gap Extension Penalty : Protein weight matrix :Gonnet series matrice de scores donnant la similarité des acides aminés les par rapport aux autres. On peut choisir entre BLOSUM 30,PAM 35, Gonnet 250, la matrice identité ou une matrice personnelle. 4. DNA weight matrix :IUB matrice de scores pour les acides nucléiques. Fast/Approximate alignments: Si on augmente la taille des k-tuples, la vitesse augmente tandis que si on la diminue, c est la précision qui augmente. [les maximum sont de 2 pour les protéines et 4 pour les acides nucléiques] 5. Gap penalty :3 cela n a que peut d influence sur la rapidité de l alignement sauf pour des valeurs extrêmes. 6. K-tuple (word) size :1 7. No. of top diagonals :5 nombre de k-tuples qui matchent sur chaque diagonale (dans un dotplot imaginaire) et seuls les meilleurs sont utilisés pour l alignement. Une diminution de ce paramètre rend l alignement plus rapide tandis qu une augmentation améliore la sensibilité. 8. Window size :5. 9. Toggle Slow/Fast pairwise alignments = SLOW.")
141
********* MULTIPLE ALIGNMENT PARAMETERS *********
1. Gap Opening Penalty : 2. Gap Extension Penalty :0.20 3. Delay divergent sequences :30 % donne le seuil au-dessus duquel l'alignement est retardé : ainsi, si une séquence donnée est plus de 30% différentes des autres, son alignement est remis à plus tard 4. DNA Transitions Weight :0.50 (poids des transitions A <-> G, C <-> T) : Un poids de zéro signifie que les transitions seront considérées comme des mismatches. Pour des séquences éloignées, ce poids doit être proche de zéro tandis qu'il est égal à 1 pour des séquences très proches 5. Protein weight matrix :Gonnet series 6. DNA weight matrix :IUB 7. Use negative matrix :OFF 8. Protein Gap Parameters
 : Un poids. de zéro signifie que les transitions seront considérées comme des mismatches. Pour des séquences éloignées, ce poids doit être proche de zéro tandis qu il est égal à 1. pour des séquences très proches. 5. Protein weight matrix :Gonnet series 6. DNA weight matrix :IUB 7. Use negative matrix :OFF. 8. Protein Gap Parameters.")
142
********* PROTEIN GAP PARAMETERS *********
1. Toggle Residue-Specific Penalties :ON Ce sont des pénalités sur certains acides aminés. Ces pénalités augment ou diminuent la probabilité d'ouverture d'un gap selon la position sur la séquence. Par exemple, les positions riches en glycine seront plus souvent adjacentes à un gap que les positions riches en valine 2. Toggle Hydrophilic Penalties :ON augmente la probabilité d'avoir des gaps dans les régions hydrophiles correspondant souvent à des boucles ou des coils 3. Hydrophilic Residues :GPSNDQEKR 4. Gap Separation Distance :4 ce paramètre tente de diminuer les risques d'avoir des gaps trop proches les uns des autres. Les gaps qui sont plus proches que cette distance sont plus pénalisés que les autres. Cela n'empêche pas d'avoir des gaps très proches, cela les rend seulement moins fréquents, donnant une apparence de blocs à l'alignement. 5. Toggle End Gap Separation :OFF Si ce paramètre est sur OFF, les gaps en fin de séquences sont ignorés, ce qui est utile lorsque l'on veut aligner des fragments où les gaps terminaux n'ont pas de signification biologique.

143
****** PROFILE AND STRUCTURE ALIGNMENT MENU ******
1. Input 1st. profile 2. Input 2nd. profile/sequences 3. Align 2nd. profile to 1st. profile permet d'aligner deux alignements déjà existants (même si chacun d'eux ne contient qu'une séquence) 4. Align sequences to 1st. profile (Slow/Accurate) permet d'ajouter une (ou une série de ) nouvelle séquence à un alignement déjà existant. L'intérêt de ce choix est double : - on peut ainsi construire un alignement de façon progressive en ajoutant de nouvelles séquences (souvent, seul un petit nombre de séquences sont à l'origine de nombreux mésappariements : on pourra les ajouter seulement à la fin) on peut avoir un alignement de référence sur lequel on aligne les nouvelles séquences. 5. Toggle Slow/Fast pairwise alignments = SLOW 6. Pairwise alignment parameters 7. Multiple alignment parameters 8. Toggle screen display = ON 9. Output format options 0. Secondary structure options
 4. Align sequences to 1st. profile (Slow/Accurate) permet d ajouter une (ou une série de ) nouvelle séquence à un alignement déjà existant. L intérêt de ce choix est double : - on peut ainsi construire un alignement de façon progressive en ajoutant. de nouvelles séquences (souvent, seul un petit nombre de séquences sont à. l origine de nombreux mésappariements : on pourra les ajouter seulement à. la fin). - on peut avoir un alignement de référence sur lequel on aligne les nouvelles. séquences. 5. Toggle Slow/Fast pairwise alignments = SLOW. 6. Pairwise alignment parameters 7. Multiple alignment parameters. 8. Toggle screen display = ON 9. Output format options 0. Secondary structure options.")
144
********* SECONDARY STRUCTURE OPTIONS *********
1. Use profile 1 secondary structure / penalty mask = YES 2. Use profile 2 secondary structure / penalty mask = YES 3. Output in alignment = Secondary Structure 4. Helix gap penalty :4 5. Strand gap penalty :4 6. Loop gap penalty :1 7. Secondary structure terminal penalty :2 8. Helix terminal positions within :3 outside :0 9. Strand terminal positions within :1 outside :1 Si une structure secondaire existe elle peut être utilisée Pour guider l’alignement

145
PHYLOGENETIC TREE MENU
****** PHYLOGENETIC TREE MENU ****** La méthode utilisée est la méthode du Neigbour-Joining (NJ) développée par Saitou et Nei : on calcule d'abord la distance (proportionnelle à la divergence) entre toutes les paires de séquences de l'alignement puis on applique la méthode de NJ sur la matrice de distance. 1. Input an alignment 2. Exclude positions with gaps? = OFF si ce paramètre est sur ON, alors toutes les positions auxquelles n'importe laquelle des séquences possède un gaps seront ignorées : cela a pour conséquence de perdre une grande quantité d'informations si l'alignement contient de nombreux gaps. 3. Correct for multiple substitutions? = OFF pour des séquences faiblement divergentes (<10%), cette option n'a pas d'intérêt. pour des divergences plus importante, cela corrige le fait que les distances observées sous-estiment les distances évolutives. Cette option à pour effet de raccourcir les longues branches d'un arbre et doit toujours être utilisée mais il faut savoir que pour des séquences très divergentes, les distances ne peuvent pas être relier de façon convenable 4. Draw tree now 5. Bootstrap tree cette méthode statistique permet d'estimer la confiance que l'on peut avoir dans l'arbre obtenu. Cette méthode génère un échantillonnage aléatoire à partir des données initiales puis compte combien de fois chaque regroupement de l'arbre initial se retrouve dans l'échantillonnage. 6. Output format options
 développée par Saitou. et Nei : on calcule d abord la distance (proportionnelle à la divergence) entre toutes. les paires de séquences de l alignement puis on applique la méthode de NJ sur la. matrice de distance. 1. Input an alignment 2. Exclude positions with gaps = OFF si ce paramètre est sur ON, alors. toutes les positions auxquelles n importe laquelle des séquences possède un. gaps seront ignorées : cela a pour conséquence de perdre une grande. quantité d informations si l alignement contient de nombreux gaps. 3. Correct for multiple substitutions = OFF pour des séquences faiblement. divergentes (<10%), cette option n a pas d intérêt. pour des divergences plus. importante, cela corrige le fait que les distances observées sous-estiment les. distances évolutives. Cette option à pour effet de raccourcir les longues. branches d un arbre et doit toujours être utilisée mais il faut savoir que pour. des séquences très divergentes, les distances ne peuvent pas être relier de. façon convenable 4. Draw tree now 5. Bootstrap tree cette méthode statistique permet d estimer la confiance que l on peut avoir dans l arbre obtenu. Cette méthode génère un échantillonnage aléatoire à partir des données initiales puis compte combien de fois chaque regroupement de l arbre initial se retrouve dans l échantillonnage. 6. Output format options.")
146
Recherche de similarités entre séquences biologiques
Clustalw La comparaison simultanée de plusieurs séquences est un outil très utile pour mieux comprendre la structure et l'évolution des protéines et des acides nucléiques mais il faut rester critique au niveau des résultats : l'alignement optimal calculé par ordinateur est rarement le meilleur au sens biologique. Il faut toujours vérifier un alignement avant de passer à l'étape suivante (phylogénie par exemple) et il peut être nécessaire de le corriger. Il faut également savoir que l'ordre des séquences dans le fichier d'entrée joue un rôle important.
 et il peut être nécessaire de le corriger. Il faut également savoir que l ordre des séquences dans le fichier d entrée joue un rôle important.")
Présentations similaires
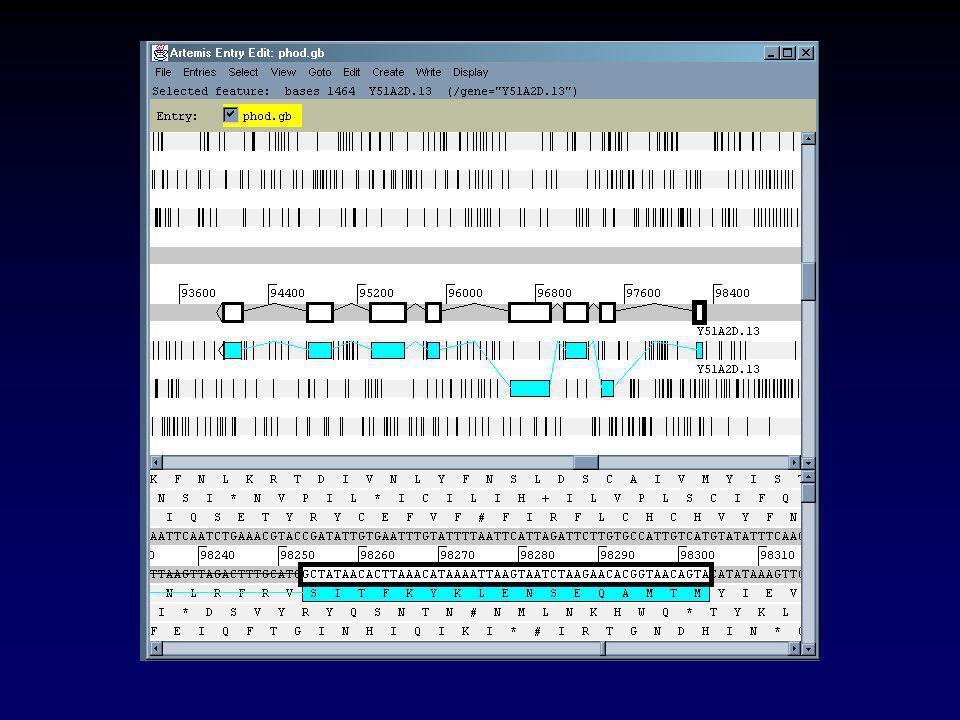
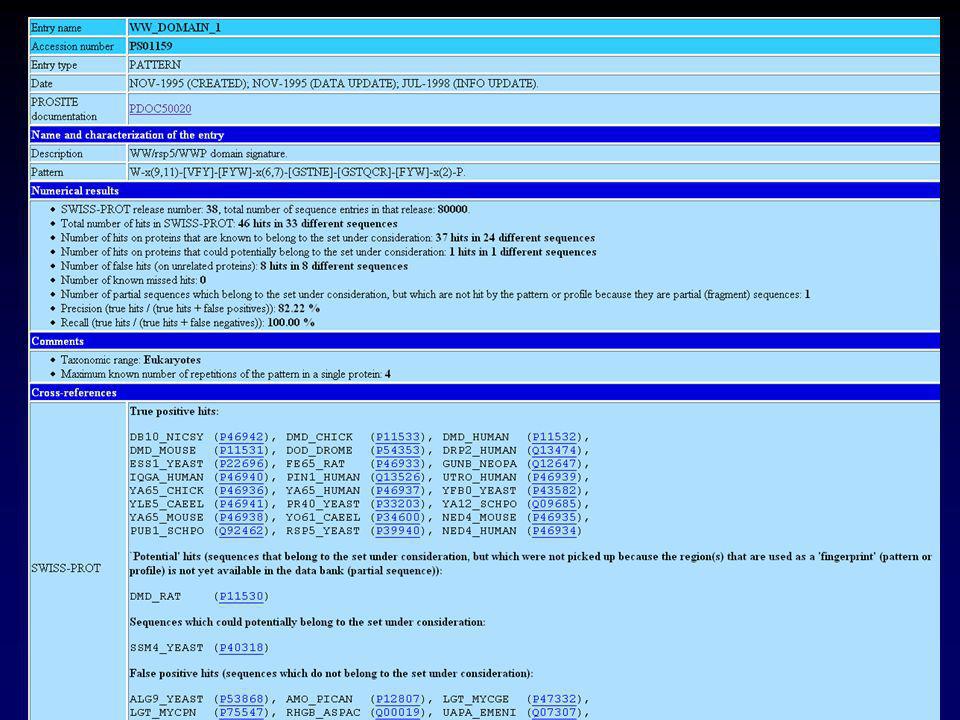

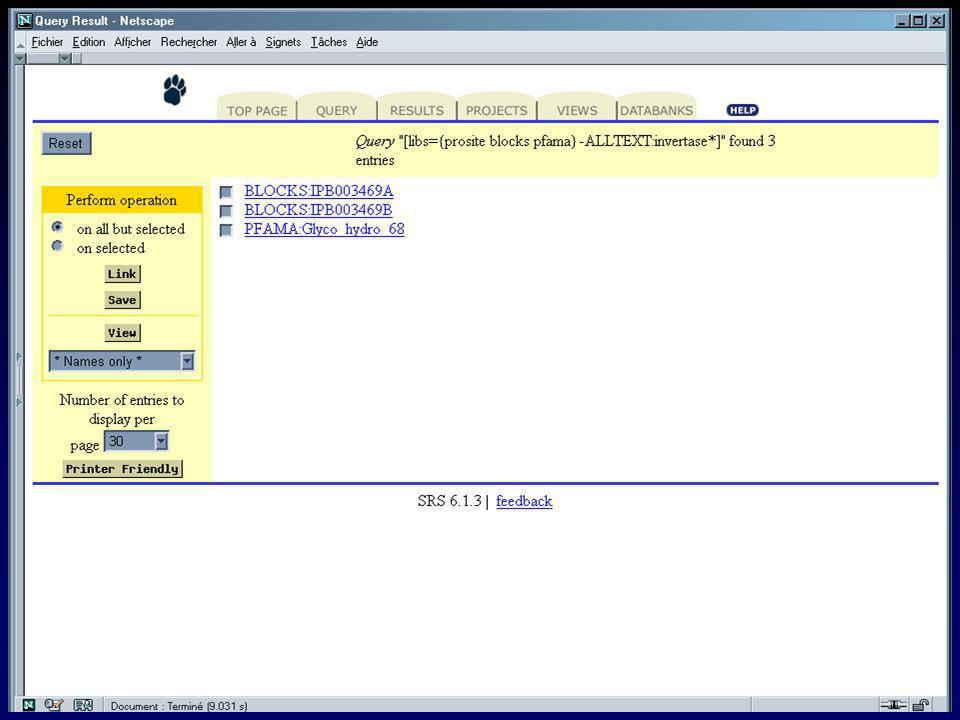
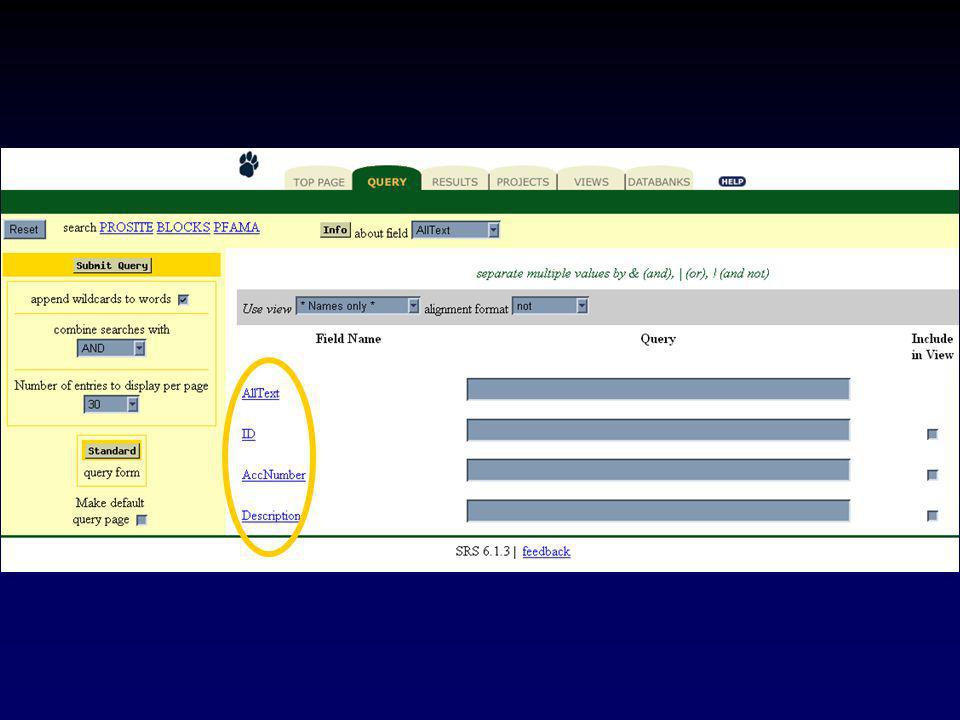
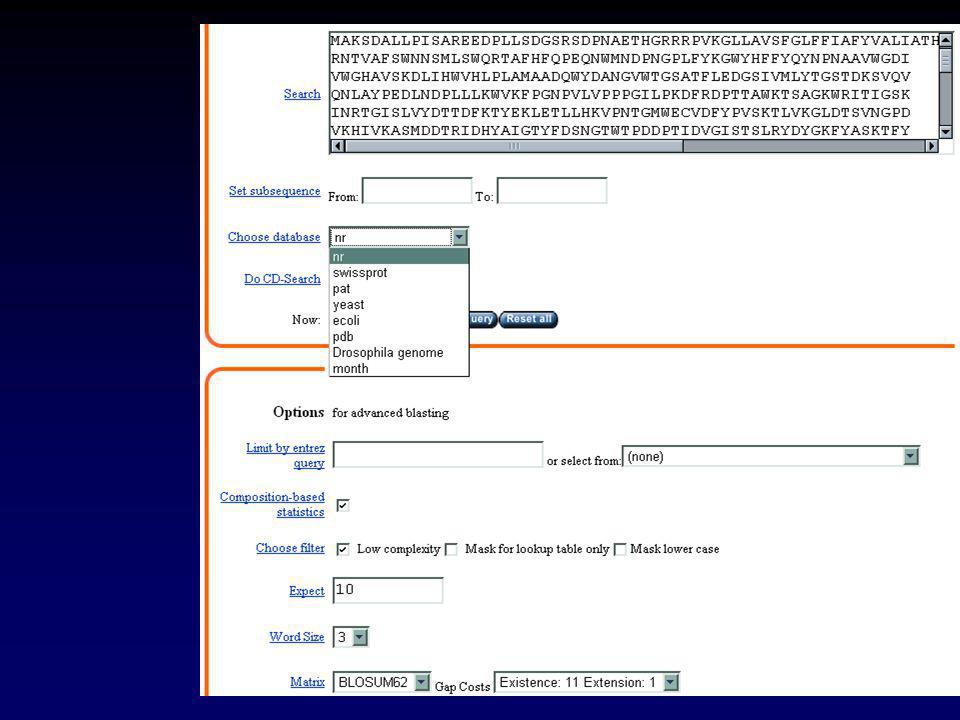
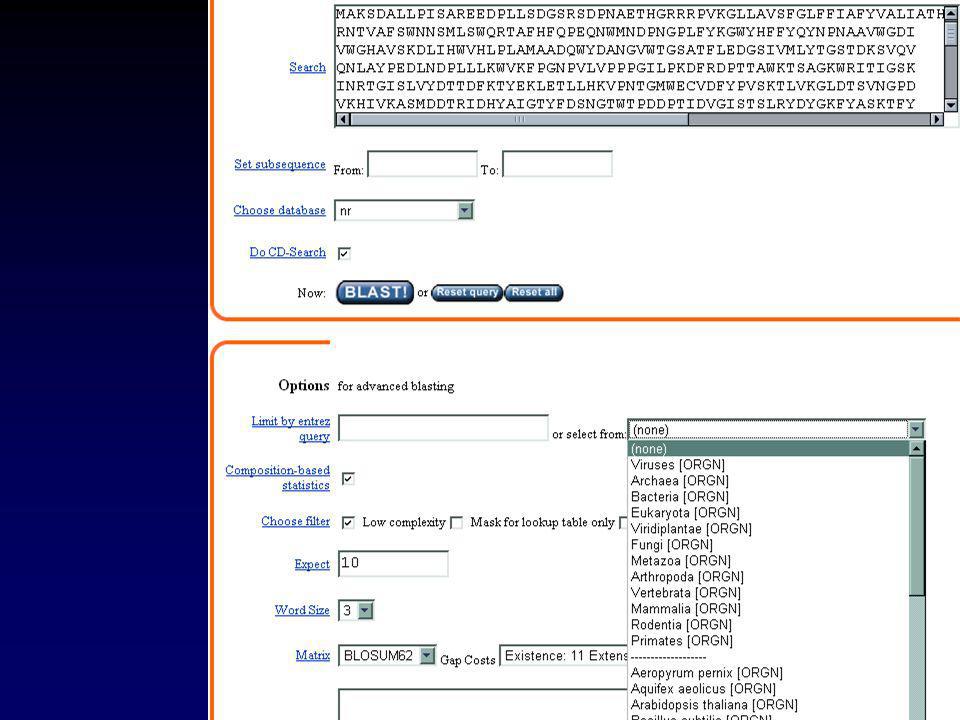
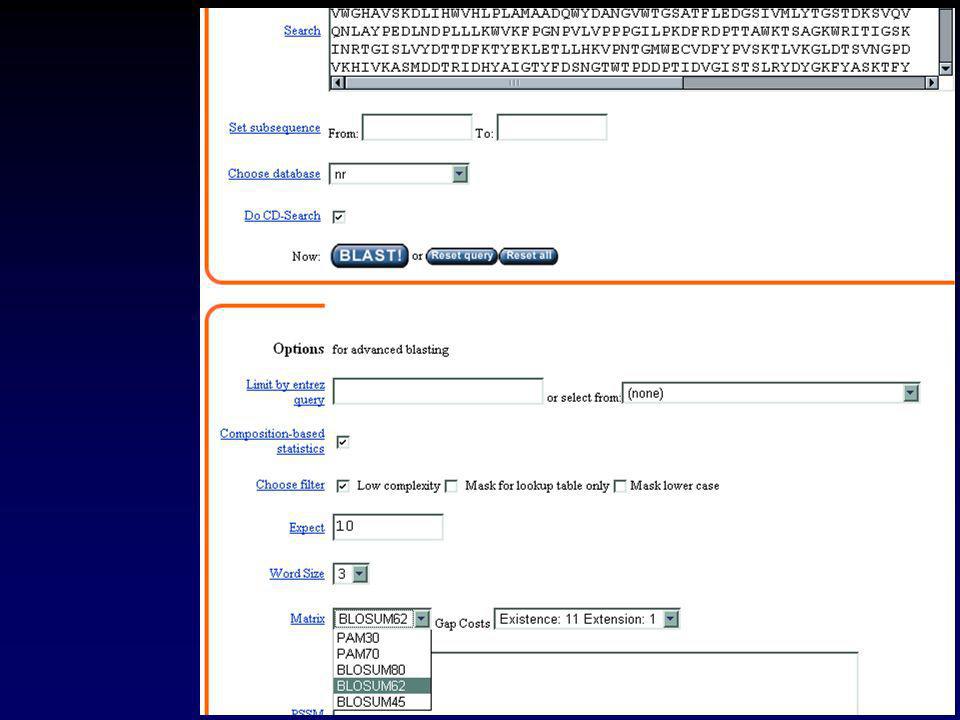

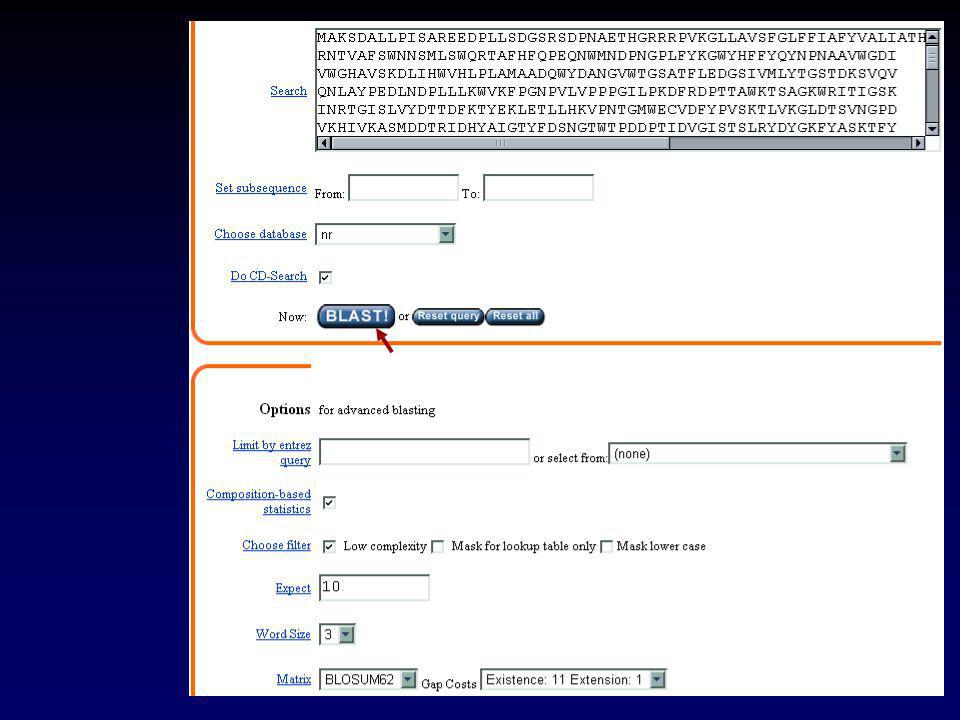










 Obtention de l’ADN recombinant>")
 polymorphes (entre individus, espèces, …) permettant - l’établissement de cartes.>")



 Phosphatases: retirent un phosphate.>")



>")
