Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Maladies héréditaires du métabolisme chez l’adulte
F Maillot Service de médecine interne et nutrition CHU de Tours

2
MHM? E = Erreurs innées du métabolisme (EIM)
Toutes ces pathologies ont en commun un défaut d’action enzymatique (voies de synthèse ou de dégradation) d’origine génétique
 d’origine génétique.")
3
Problématique générale de la prise en charge des adultes avec EIM
Relais de la prise en charge pédiatrique Diagnostics d’EIM chez l’adulte

4
Relais de la prise en charge pédiatrique
Progrès dans la prise en charge globale Régimes spéciaux Médicaments, enzymothérapie Prise en charge psycho-sociale Amélioration de la survie des enfants atteints

5
Amélioration de la survie
population d’adolescents et jeunes adultes à prendre en charge : Complications spécifiques : ex GSD1 Aspects socioprofessionnels Contraception Grossesse (ex : PKU) et postpartum (UCD) cas particulier : interruption du suivi pédiatrique
 et postpartum (UCD) cas particulier : interruption du suivi pédiatrique.")
6
Glycogénoses hépatiques chez l’adulte

7
Glycogénoses hépatiques (n=260)
type enzyme répartition Glycogène Synthase ? I Glucose-6-Phosphatase 27 % III Enzyme débranchante 28 % IV Enzyme branchante 2 % VI Phosphorylase 7 % VIII Phosphorylase kinase 33 % XI GLUT 2 3 % Autres Glycogénoses : Type II = Maladie de Pompe, Type V = Mc Ardle, …

8
VIII Glycogène VI IV III Glucose-1-P Glucose-6-P Glucose I K Ch Respi
G-1-P VIII Glycogène VI IV III Glucose Glucose-1-P Glucose-6-P Glucose I K Ch Respi Pyruvate Lactate FOIE SANG

9
GSD1 Glycogène Glycogène = Gros foie Glucose-1-P Acide urique
G-6-Phosphatase Foie, rein Glycogène Glycogène = Gros foie Glucose-1-P Acide urique Glucose-6-P Glucose I Triglycérides K Ch Respi Pyruvate Lactate FOIE SANG

10
GSD1 chez l’enfant Clinique : Biologie :
Gros foie (avec adénomes), obésité facio-tronculaire Hypoglycémie pour un jeûne court Atteinte rénale (tubulopathie) Petite taille …. Biologie : Jeûne court : hypoglycémie + hyperlactacidémie HyperTG, hyperuricémie Autres : Transa , tubulopathie, rachitisme…
, obésité facio-tronculaire. Hypoglycémie pour un jeûne court. Atteinte rénale (tubulopathie) Petite taille. …. Biologie : Jeûne court : hypoglycémie + hyperlactacidémie. HyperTG, hyperuricémie. Autres : Transa , tubulopathie, rachitisme…")
11
GSD1 chez l’adulte Même tableau clinique, dont la sévérité dépend de l’équilibre métabolique, avec une amélioration de la tolérance au jeûne Complications spécifiques : Glomérulopathie, insuffisance rénale Ostéopénie Hépatocarcinome

12
GSD1 - hépatocarcinome Cohorte ESGSD 1 (n = 288)
Aucun cas de « transformation » d’adénome en hépatocarcicome (Rake JP et al, Eur J Pediatr 2002) Adultes : 8 cas âgés de 39 ± 9,9 ans Mauvaise compliance thérapeutique Suivi irrégulier Faibles sensibilités de L’AFP et échographie Franco et al, JIMD 2005
 Adultes : 8 cas âgés de 39 ± 9,9 ans. Mauvaise compliance thérapeutique. Suivi irrégulier. Faibles sensibilités de L’AFP et échographie. Franco et al, JIMD")
13
La phénylcétonurie chez l’adulte

14
Relais de la prise en charge pédiatrique
2 problèmes chez l’adulte: 1) maintien du régime pauvre en Phe? 2) Grossesses des femmes PCU
 maintien du régime pauvre en Phe 2) Grossesses des femmes PCU.")
15
Maintien du régime ? État des lieux
50 à 90% des adultes avec PCU classique ne le suivent plus Parmi ceux qui le suivent: + de 70% ont une Phe supérieure au taux recommandé Taux recommandé variable*: < 600 µmol/L (USA), < 700 (UK), < 1200 (Allemagne), < 1300 (France)! *Hanley WB, Am J Med 2004
, < 700 (UK), < 1200 (Allemagne), < 1300 (France)! *Hanley WB, Am J Med")
16
Régime pour la vie ? Arguments pour
Observations : détérioration neurologique difficultés d’apprentissage troubles neuropsychologiques problèmes psychiatriques et psychosociaux Souhait de certains patients de poursuivre le régime Préparation des patientes vis-à-vis des grossesses

17
Maintien du régime, oui mais..
Anomalies aux tests neuropsychologiques : traduction clinique? Intérêt du régime restrictif non démontré par des études à long terme Le régime est difficile à suivre (surtout s’il a été interrompu) pour un adulte, et d’autres traitements seront prochainement disponibles Walter et al., Lancet 2002
 pour un adulte, et d’autres traitements seront prochainement disponibles. Walter et al., Lancet")
18
Grossesse d’une patiente PCU
Problème délicat de la toxicité de l’hyperphénylalaninémie sur le fœtus microcéphalie Retard de croissance intra-utérin Malformations cardiaques Retard mental Nécessité d’un contrôle métabolique très strict, qu’il faut obtenir avant la grossesse et pendant toute sa durée

19
Grossesse Régime pauvre en Phe : même principe que chez l’enfant, mais: La reprise du régime est difficile chez un adulte qui l’a abandonné Il faut tenir compte des besoins nutritionnels spécifiques de la femme enceinte, et compenser les éventuelles carences La patiente doit avoir été informée avant la grossesse des risques et des modalités de prise en charge Objectif métabolique: Phe µmol/L

20
PCU et grossesse en France
Etude rétrospective de 135 grossesses (2001) * 79 patientes Diagnostic: 74% par dépistage NN, 18% enfance, 8% après embryopathie (un cas de ce type observé à Tours) 73% avec PCU classique Feillet et al., Eur J Pediatr 2004
 * 79 patientes. Diagnostic: 74% par dépistage NN, 18% enfance, 8% après embryopathie (un cas de ce type observé à Tours) 73% avec PCU classique. Feillet et al., Eur J Pediatr")
21
PCU et grossesse en France
36% des patientes n’étaient pas informés des risques (DN < 1970 – DN > 1978: 80% informées) Cf Me M…, CHU de Tours, ITG pour microcéphalie grave, alors que la PCU était connue..seulement de sa mère! Régime pré-conceptionnel : 57/135 grossesses (42,2%)
 Cf Me M…, CHU de Tours, ITG pour microcéphalie grave, alors que la PCU était connue..seulement de sa mère! Régime pré-conceptionnel : 57/135 grossesses (42,2%)")
22
Pronostic des grossesses PCU (n=124)
Avortements spontanés 10,4% Avortements thérapeutiques 12,6% Embryopathie 21,2% (6 > diagnostic PCU). Incidence en diminution, aucun cas après 2001. 43% des enfants en bonne santé, % en augmentation avec le temps Mères informées : 80% d’enfants en bonne santé
. Incidence en diminution, aucun cas après % des enfants en bonne santé, % en augmentation avec le temps. Mères informées : 80% d’enfants en bonne santé.")
23
PCU chez l’adulte: conclusions
L’intérêt du « régime à vie » doit être démontré Importance de l’information des patientes vis-à-vis des risques d’une grossesse sans régime Nécessité de développer la prise en charge des patients adultes PCU, pour: Surveiller les adultes au long cours (avec ou sans régime) Améliorer le pronostic des grossesses
 Améliorer le pronostic des grossesses.")
24
Le relais de la prise en charge pédiatrique ne se limite pas à ces deux exemples :
Acidémies organiques Leucinose Galactosémie GSD III UCD ….

25
Diagnostics à l’âge adulte
Maladies classiquement diagnostiquées chez l’adulte M de Gaucher, Fabry, porphyrie aigue Formes « atténuées » de maladies classiquement diagnostiquées chez l’enfant UCD, homocystinurie, glycogénoses

26
Diagnostic des IEM en France 1997/98

27
Tableau clinique, Diagnostic
Maladie de Fabry Tableau clinique, Diagnostic

28
Définition Maladie de surcharge du lysosome : sphingolipidose
Incidence :1 naissance masculine / Mode de transmission récessif lié à l’X Déficit enzymatique en -galactosidase A

29
Physiopathologie Accumulation progressive de glycosphingolipides, principalement le globotriaosylcéramide (GL-3) dans les lysosomes d’un grand nombre de cellules. Accumulation modérée de GL-3 Accumulation sévère de GL-3
 dans les lysosomes d’un grand nombre de cellules. Accumulation modérée de GL-3. Accumulation sévère de GL-3.")
30
Atteintes multisystémiques :
Physiopathologie Accumulation de GL-3 dans Les cellules endothéliales, perithéliales et musculaires lisses des vaisseaux, Les cellules épithéliales de la cornée, Les cellules nerveuses des ganglions postérieurs de la moelle et du système nerveux autonome, Les cellules glomérulaires et tubulaires du rein, Les cardiomyocytes, Les fibrocytes valvulaires, … Atteintes multisystémiques : cutanées, oculaires, auditives, rénales, cardiaques et neurologiques

31
Enfance Acroparesthésies et douleurs intermittentes (arthralgies)
Hypo/anhidrose Asthénie et fièvres récidiventes Atteinte rénale souvent infraclinique à ce stade Opacités cornéennes (cornée verticillée) sans atteinte de l’acuité visuelle
 sans atteinte. de l’acuité visuelle.")
32
Adolescence Acroparesthésies et douleurs intermittentes (arthralgies)
Hypo/anhidrose Asthénie et fièvre Atteinte rénale souvent infraclinique à ce stade Opacités cornéennes (cornée verticillée) sans atteinte de l’acuité visuelle Syndrome digestif Angiokératomes Répercussions psychosociales
 sans atteinte. de l’acuité visuelle. Syndrome digestif. Angiokératomes. Répercussions psychosociales.")
33
Adulte Syndrome digestif Angiokératomes Répercussions psychosociales
Hypo/anhidrose Asthénie et fièvre Opacités cornéennes (cornée verticillée) sans atteinte de l’acuité visuelle Insuffisance rénale grave puis terminale Troubles du SNC (AIT, AVC précoces) Atteintes cardiaques Hypoacousie, voire surdité
 sans atteinte. de l’acuité visuelle. Insuffisance rénale grave puis terminale. Troubles du SNC (AIT, AVC précoces) Atteintes cardiaques. Hypoacousie, voire surdité.")
34
Oeil Cornée verticillée et atteintes cristalliniennes postérieures, lésions angiomateuses des vaisseaux de la conjonctive, occlusion des vaisseaux rétiniens sans atteinte de l’acuité visuelle. Avec l‘aimable autorisation de R. J. Desnick, PhD, MD, New York.

35
Peau Angiokératome Avec l‘aimable autorisation de R. J. Desnick, PhD, MD, New York.

36
Rein Microalbuminurie, protéinurie, déclin de la fonction rénale jusqu’à une insuffisance rénale terminale Rein d’un patient Fabry à un stade avancé de la maladie Formation de cicatrices et kystes Gauche : vue externe, droite : vue en coupe Avec l‘aimable autorisation de E. Gilbert-Barness et L. Barness

37
Diagnostic Mesure de l’activité enzymatique de l’-galactosidase A dans les leucocytes (85% des patients ont une activité nulle), taux normal: 25 à 55 nmol/h/mg de protéine. Augmentation des taux de GL-3 plasmatique et urinaire Enquête familiale Diagnostic génétique (génotypage, recherche d’une mutation du gène GLA)
, taux normal: 25 à 55 nmol/h/mg de protéine. Augmentation des taux de GL-3 plasmatique et urinaire. Enquête familiale. Diagnostic génétique (génotypage, recherche d’une mutation du gène GLA)")
38
Age au diagnostic et sévérité
Atteinte cardiaque Atteinte du SNC Atteinte rénale Acroparesthésies Angiokératome Non diagnostiqué 30 60 (ans) Âge moyen du diagnostic: 29 ans Diagnostiqué peu sévère Diagnostiqué sévère
 Âge moyen du diagnostic: 29 ans. Diagnostiqué. peu sévère. Diagnostiqué sévère.")
39
Maladie de Fabry conclusions
Maladie évolutive de l'enfance à l’âge adulte d’aggravation constante Enzymothérapie disponible : intérêt du diagnostic précoce

40
Cycle de l'urée CAA plasma et urines Acide orotique urinaire
Glutamate N-Acetyl-Glutamate NH 4 + Carbamylphosphate Citrulline Argininosuccinate Arginine Ornithine MITOCHONDRIE CYTOSOL UREE Acetyl-CoA Acide orotique Œ ‘ Diagnostic biochimique CAA plasma et urines Acide orotique urinaire

41
UCD : Observation Mme BEL.. K, 19 ans antécédents = 0
pas de traitement Un mois avant l ’hospitalisation douleurs hypochondre droit et nausées NFS, glycémie normaux écho abdominale normale traitement par Primpéran

42
UCD : cas clinique Mme BEL.. K, apparition progressive de
Signes neurologiques acouphènes, troubles de l’humeur, épisodes de somnolence, troubles de l ’élocution Signes digestifs douleurs abdominales, vomissements Scanner cérébral (10/8/00) : normal
 : normal.")
43
Appel du SMUR (dimanche 13/8/00)
état stuporeux Aux urgences alternance mutisme total, obnubilation agitation, gestes incohérents, désinhibition absence de signe méningé signes déficitaires fièvre abdomen souple

44
screening toxicologique négatif PL Prot = 0,35 g/l Gly = 4 mmol/l
Biologie usuelle Biologie spécialisée screening toxicologique négatif PL Prot = 0,35 g/l Gly = 4 mmol/l cel = 2/mm3 EEG Hospitalisation Souffrance cérébrale diffuse

45
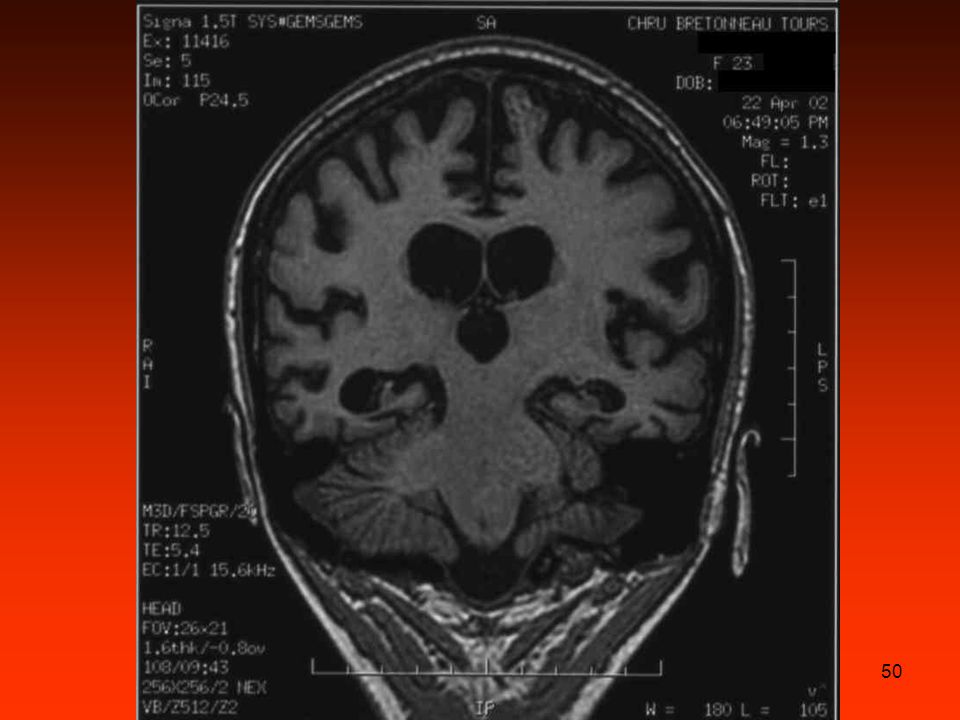
coma réanimation, intubation coma calme ; CGS = 6/15 pas de fièvre
14/8/00 : aggravation coma réanimation, intubation coma calme ; CGS = 6/15 pas de fièvre IRM en urgence normale Interrogatoire de la famille : cousine décédée 3 ans plus tôt, dans des circonstances identiques au cas actuel +++ Cause métabolique rare ??

46
Dosages urinaires en urgence
Porphyrie aiguë ?? Dosages urinaires en urgence acide delta aminolévulinique = 9 µmol/l (N<38) porphobilinogène = 2 µmol/l (N <5) 15/8/00 : ammoniémie = 606 µmol/l état de mal convulsif scanner cérébral: œdème cérébral majeur
 porphobilinogène = 2 µmol/l (N <5) 15/8/00 : ammoniémie = 606 µmol/l. état de mal convulsif. scanner cérébral: œdème cérébral majeur.")
47
OCT Acide orotique

48
Traitement spécifique :
nutrition hypercalorique aprotidique médicaments « épurateurs » HDF continue Traitement symptomatique : état de mal épileptique œdème cérébral

49
Évolution neurologique HTIC non contrôlée
ammoniémie HDF - métabolique Évolution neurologique HTIC non contrôlée engagements cérébraux pluriquotidiens DECES LE 23/8/00 (J10)
")
51
UCD adultes : série française
12 patients (9 F et 3 H), âgés de 20 à 58 ans lors du diagnostic Déficit OTC CPS ASL n forme aiguë forme chronique
, âgés de 20 à 58 ans lors du diagnostic. Déficit OTC CPS ASL. n forme aiguë forme chronique")
52
UCD adultes 1987 1996 1987 1998 2000 2000 2000 2000 2002 ? 2000 1993 2002

53
Conclusions La médecine d’adultes (internistes, neurologues,…) a sa place dans la prise en charge des EIM le nombre d’enfants qui survivent et atteignent l’âge adulte augmente Les formes cliniques de l’adulte sont mieux connues Un effort d’organisation est nécessaire : activité disséminée, sauf pour les maladies lysosomales (CETL), Wilson,….
, Wilson,….")
54
Activité Adulte, CHU de Tours
Nbre Patients/an 13 19 31 Hospit (j) Cs (nbre)
 Cs (nbre)")
56
Remerciements Dr François Labarthe, service de Pédiatrie R, CHU de Tours (tableaux et schémas GSD1) Dr Olivier Brenet, service de réanimation, CH de Cholet (observation d ’UCD)
")
Présentations similaires