Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Faculté de Pharmacie de Monastir
Année Universitaire Cours Hémostase: 3ème Année Pharmacie LA COAGULATION - Physiologie et exploration - Principales maladies hémorragiques L’hémophilie - Les déficits congénitaux en facteurs de la coagulation Pr. NSIRI B

2
L’hémostase: ensemble des mécanismes qui concourent à:
INTRODUCTION L’hémostase: ensemble des mécanismes qui concourent à: - Arrêt des hémorragies - Prévention des saignements spontanés - Prévention des thromboses Elle participe à la réparation de la brèche vasculaire et assure le maintien de l’intégrité des vaisseaux..

3
INTRODUCTION Série de systèmes en équilibre qui sont dépendants
l’un de l’autre: - Système vasculaire - Système plaquettaire - Système de la coagulation - Système de la fibrinolyse

4
INTRODUCTION Thrombose Hémorragie
Elle est souvent comparée à une balance à l’état d’équilibre: Toute rupture de l’équilibre fera pencher la balance soit vers un état thrombotique soit vers un état hémorragique Thrombose Hémorragie

5
LES ETAPES DE L’HEMOSTASE
Schématiquement, l’hémostase comporte 3 temps: - L’hémostase primaire - La coagulation - La fibrinolyse

6
LES ETAPES DE L’HEMOSTASE
Hémostase primaire: interactions plaquettes-vaisseaux: Clou plaquettaire ou thrombus blanc Coagulation: transformation du fibrinogène en fibrine: Caillot de fibrine ou thrombus rouge Fibrinolyse: transformation du plasminogène en plasmine: Disparition du caillot et cicatrisation des vaisseaux

7
PHYSIOLOGIE DE LA COAGULATION
La coagulation se fait par une cascade de réactions enzymatiques aboutissant à la transformation du fibrinogène soluble en un gel de Fibrine insoluble. La coagulation ne peut se dérouler sans la présence de cellules ou de substances originaires de ces cellules. Les cellules les plus importantes dans la coagulation sont les cellules endothéliales, les monocytes et les plaquettes. Les cellules endothéliales stimulées par des cytokines ou des facteurs physico-chimiques, expriment à leur surface une protéine, le facteur tissulaire, qui est associée à des phospholipides membranaires. Ce facteur tissulaire anciennement appelé thromboplastine tissulaire est l'élément déclenchant et le support majeur de la coagulation. Des cellules circulantes, les monocytes, sont également capables d'exprimer le facteur tissulaire sous l'influence de cytokines (IL1, TNF) voire d'endotoxine bactérienne ou de certains antigènes. Les plaquettes interviennent aussi dans la coagulation. Lorsque la plaquette est activée, les phospholipides anioniques membranaires sont externalisés et serviront de support à la coagulation Enfin les plaquettes (tout comme les monocytes) peuvent libérer dans le milieu plasmatique de petits fragments de membrane appelés microvésicules capables elles aussi de supporter le phénomène de coagulation et donc de l'amplifier. D'autres cellules peuvent jouer un rôle dans la coagulation : les fibroblastes sont capables d'exprimer le facteur tissulaire; ils synthétisent tout comme les cellules musculaires beaucoup de facteurs impliqués dans la coagulation.
 voire d endotoxine bactérienne ou de certains antigènes. Les plaquettes interviennent aussi dans la coagulation. Lorsque la plaquette est activée, les phospholipides anioniques membranaires sont externalisés et serviront de support à la coagulation Enfin les plaquettes (tout comme les monocytes) peuvent libérer dans le milieu plasmatique de petits fragments de membrane appelés microvésicules capables elles aussi de supporter le phénomène de coagulation et donc de l amplifier. D autres cellules peuvent jouer un rôle dans la coagulation : les fibroblastes sont capables d exprimer le facteur tissulaire; ils synthétisent tout comme les cellules musculaires beaucoup de facteurs impliqués dans la coagulation.")
8
I- Paramètres impliqués dans la coagulation
- Phospholipides membranaires anioniques - D’origine cellulaire: plaquette activée, cellule endothéliale, monocyte.. - D’origine tissulaire: Facteur tissulaire ( FT) - Protéines plasmatiques activateurs et inhibiteurs de la coagulation - ions calcium Assurent les liens entre les PL et les facteurs de la coagulation vitamine K dépendants et du FXI
 - Protéines plasmatiques activateurs et inhibiteurs de la coagulation - ions calcium Assurent les liens entre les PL et les facteurs de la coagulation vitamine K dépendants et du FXI")
9
Protéines plasmatiques (Facteurs) de la coagulation
Lieu de synthèse Dénomination Facteurs Foie Fibrinogène I Foie + Vit K dépendant Prothrombine II Proaccélérine V Proconvertine VII Foie, cellule endothéliale F. anti hémophilique A VIII F. anti hémophilique B IX F. Stuart X F. Rosenthal XI F. Hageman XII Foie, plaquette, monocyte F. stabilisant de la Fb XIII 2 - Eléments non cellulaires: facteurs de coagulation et leurs inhibiteurs Les facteurs de coagulation sont des pro-enzymes toutes synthétisés par l'hépatocyte. Ceci explique les désordres hémorragiques chez les cirrhotiques ou les personnes atteintes d'une insuffisance hépato-cellulaire. Le facteur VIII fait exception à cette règle : son taux reste normal ou augmenté. Il existe toujours au moins 2 formes pour ces facteurs: une forme non active (exemple facteur VII: proconvertine, facteur II: prothrombine) et une forme active (exemple facteur VIIa: convertine, facteur IIa: thrombine). Aucun de ces facteurs n'a de substrat spécifique. Chaque facteur à l'état activé pourra soit activer un autre facteur soit modifier certaines protéines impliquées ou non dans la coagulation. Le seul substrat vrai de la coagulation sera en fait le fibrinogène. L’ensemble de ces facteurs est repris dans le tableau 1. Certains de ces facteurs portent des résidus gamma-carboxylés qui leur permettent de fixer le calcium et de se lier aux membranes phospholipidiques. Il s'agit des facteurs II, VII, X, IX (habituellement désignés par PPSB du nom de leurs initiales : Prothrombine, Proconvertine, facteur Stuart, facteur antihémophilique B), et de certains inhibiteurs: protéine C, protéine S. La Gamma-carboxylation nécessite la présence de vitamine K d'où le nom de facteur vitamine K dépendant. Ainsi, un patient porteur d'une avitaminose K ou recevant un traitement appelé antivitamine K aura une diminution de synthèse de ces facteurs. A la place circulent des substances appelées PIVKA (Protein Induced by Vitamine K Absence ou Antagoniste): PIVKA VII, PIVKA II, PIVKA X, PIVKA IX : ce sont des précurseurs non carboxylés donc inactifs car leur liaison aux phospholipides en présence de calcium est impossible.
 et une forme active (exemple facteur VIIa: convertine, facteur IIa: thrombine). Aucun de ces facteurs n a de substrat spécifique. Chaque facteur à l état activé pourra soit activer un autre facteur soit modifier certaines protéines impliquées ou non dans la coagulation. Le seul substrat vrai de la coagulation sera en fait le fibrinogène. L’ensemble de ces facteurs est repris dans le tableau 1. Certains de ces facteurs portent des résidus gamma-carboxylés qui leur permettent de fixer le calcium et de se lier aux membranes phospholipidiques. Il s agit des facteurs II, VII, X, IX (habituellement désignés par PPSB du nom de leurs initiales : Prothrombine, Proconvertine, facteur Stuart, facteur antihémophilique B), et de certains inhibiteurs: protéine C, protéine S. La Gamma-carboxylation nécessite la présence de vitamine K d où le nom de facteur vitamine K dépendant. Ainsi, un patient porteur d une avitaminose K ou recevant un traitement appelé antivitamine K aura une diminution de synthèse de ces facteurs. A la place circulent des substances appelées PIVKA (Protein Induced by Vitamine K Absence ou Antagoniste): PIVKA VII, PIVKA II, PIVKA X, PIVKA IX : ce sont des précurseurs non carboxylés donc inactifs car leur liaison aux phospholipides en présence de calcium est impossible.")
10
Protéines plasmatiques (Facteurs) de la coagulation
Ces facteurs sont classés en: II, VII, IX, X, XI,XII, XIII Proenzymes V, VIII Cofacteurs Fibrinogène Substrat

11
II- Mécanismes de la coagulation in vivo
Lésion de la paroi vasculaire Facteur tissulaire Activation de la coagulation Génération de la prothrombinase La coagulation est une cascade de réactions enzymatiques. L'enzyme qui permet de transformer le fibrinogène en fibrine est la thrombine. Son processus de formation est complexe. Il comprend une série d'activations enzymatiques en cascade qui surviennent à la surface des phospholipides membranaires de certaines cellules (plaquettes, cellules endothéliales, monocytes). Génération de la thrombine Fibrinogène Fibrine
. Génération de la thrombine. Fibrinogène. Fibrine.")
12
Schéma classique de la coagulation: Théorie des deux voies
Classiquement la coagulation est divisée en deux voies: - Voie intrinsèque: voie d’activation à la surface des cellules activées (plaquettes, cellule endothéliale...). - Voie extrinsèque: voie d’activation par le facteur tissulaire. Ces deux voies partagent une voie commune qui aboutit à la formation du caillot de fibrine. 1 - La conception classique du phénomène de coagulation comportait 2 voies d'activation La voie intrinsèque dans laquelle tous les éléments nécessaires de la coagulation sont présents dans le plasma sans apport extérieur. Cette voie s'active en présence de surface mouillable comme le verre. la voie extrinsèque qui pour être activée nécessite la présence d'éléments tissulaires appelés thromboplastine tissulaire. Le déroulement de la coagulation in vivo ne respecte pas cette distinction voie intrinsèque - voie extrinsèque. Cette conception duelle de la coagulation correspond en fait aux processus de coagulation in vitro et sera très utile pour l’exploration de la coagulation car la voie intrinsèque (ou endogène) et la voie extrinsèque (ou exogène) sont respectivement explorées par le temps de céphaline activée et le temps de Quick. C'est donc sur ce schéma que pourra se faire le raisonnement diagnostique d'interprétation des tests de coagulation bien que ce schéma ne correspond pas à la réalité in vivo.
. - Voie extrinsèque: voie d’activation par le facteur tissulaire. Ces deux voies partagent une voie commune qui aboutit à la formation du caillot de fibrine. 1 - La conception classique du phénomène de coagulation comportait 2 voies d activation. La voie intrinsèque dans laquelle tous les éléments nécessaires de la coagulation sont présents dans le plasma sans apport extérieur. Cette voie s active en présence de surface mouillable comme le verre. la voie extrinsèque qui pour être activée nécessite la présence d éléments tissulaires appelés thromboplastine tissulaire. Le déroulement de la coagulation in vivo ne respecte pas cette distinction voie intrinsèque - voie extrinsèque. Cette conception duelle de la coagulation correspond en fait aux processus de coagulation in vitro et sera très utile pour l’exploration de la coagulation car la voie intrinsèque (ou endogène) et la voie extrinsèque (ou exogène) sont respectivement explorées par le temps de céphaline activée et le temps de Quick. C est donc sur ce schéma que pourra se faire le raisonnement diagnostique d interprétation des tests de coagulation bien que ce schéma ne correspond pas à la réalité in vivo.")
13
Génération de la prothrombinase (voie intrinsèque)
XIIa, XIa, PK, KHPM IX IXa, VIIIa, PL, Ca2+ Tenase (dixase) Xa, Va, PL, Ca2+ X Prothrombinase Prothrombine Thrombine
 Xa, Va, PL, Ca2+ X. Prothrombinase. Prothrombine. Thrombine.")
14
Génération de la prothrombinase (voie extrinsèque)
FVIIa- FT- Ca2+ X Xa, Va, PL, Ca2+ Prothrombinase Prothrombine Thrombine

15
Schéma général (2 voies)
FVIIa - FT - Ca2+ XIIa,XIa,PK,KHPM IXa,VIIIa,PL,Ca2+ IX X Xa, Va, PL, Ca2+ Dès qu'apparaissent des traces de thrombine, le processus de coagulation s'amplifie. La thrombine casse le fibrinogène en libérant 2 petits peptides : fibrinopeptide A et fibrinopeptide B. En perdant le fibrinopeptide A puis le fibrinopeptide B, le fibrinogène devient la fibrine. Spontanément, les monomères de fibrine peuvent se polymériser et former un premier réseau ou polymère soluble de fibrine. Ce polymère est instable. Il sera stabilisé par un dernier facteur, le facteur XIII (facteur stabilisant la fibrine: FSF). Le facteur XIII crée des liaisons covalentes solides entre ces monomères de fibrine. On a alors formation d'un réseau de fibrine qui emprisonne les globules rouges : le thrombus rouge définitif est ainsi formé (Fig 5). Prothrombine Thrombine Fibrinogène Fibrine
. Le facteur XIII crée des liaisons covalentes solides entre ces monomères de fibrine. On a alors formation d un réseau de fibrine qui emprisonne les globules rouges : le thrombus rouge définitif est ainsi formé (Fig 5). Prothrombine. Thrombine. Fibrinogène. Fibrine.")
16
Anomalies de la théorie des deux voies
1- Pas de problème de saignement chez les patients déficients en FXII, PK ou KHPM, alors que le TCA est très allongé.

17
Anomalies de la théorie des deux voies
2- Les patients déficients en FVII ont généralement un problème de saignement malgré une voie intrinsèque normale.

18
3- Le FVII peut activer le FIX alors qu’ils
Anomalies de la théorie des deux voies 3- Le FVII peut activer le FIX alors qu’ils appartiennent à deux voies différentes

19
Schéma général (2 voies)
FVIIa - FT - Ca2+ XIIa,XIa,PK,KHPM IXa,VIIIa,PL,Ca2+ IX X Xa, Va, PL, Ca2+ Prothrombine Thrombine Fibrinogène Fibrine

20
Le nouveau concept de la coagulation
La coagulation peut être séparée en 4 phases: - Phase d’initiation - Phase d’amplification - Phase de propagation et de stabilisation

21
a - La phase d’initiation
- La lésion de la paroi vasculaire entraîne un Contact direct entre le sang et le FT du sous endothélium. - Le FT active le FVII, il se forme ainsi un complexe FT-FVIIa appelé: Tenase extrinsèque. - La Tenase extrinsèque fournit de faibles quantités de FIXa et de FXa. 2 - Conception actuelle de la coagulation in vivo a) Le déclenchement de la coagulation Il est admis que l'élément déclenchant de la coagulation in vivo est l'expression à la surface des cellules (monocytes, cellules endothéliales voire fibroblastes) de facteur tissulaire. Le facteur tissulaire est un récepteur membranaire de très haute affinité pour le facteur VII. Il est normalement absent de la circulation sanguine mais est exprimé au niveau des cellules musculaires lisses de la paroi vasculaire et des fibroblastes de façon constitutive. Certains tissus sont très riches en facteur tissulaire : tissu cérébral par exemple. Ainsi, Lors d'une brèche vasculaire ou lors de l'expression pathologique du facteur tissulaire par certaines cellules sanguines, ce dernier fixe le facteur VII circulant qu’il active formant un complexe: [facteur VII activé - facteur tissulaire]. Il existe une toute petite quantité préalable de facteur VII déjà activé dans le plasma mais qui en l’absence de facteur tissulaire a très peu d’activité enzymatique. A partir de la formation du complexe, deux voies d'activation sont possibles (Fig.4): Quand le facteur tissulaire est en excès, le complexe [facteur VII activé - facteur tissulaire]. active directement le facteur X. Cette voie peut cependant être rapidement inhibée par l’inhibiteur de la voie du facteur tissulaire, le TFPI. Quand le facteur tissulaire est en faible quantité (ou l'inhibition par le TFPI prépondérante), le complexe [facteur VII activé - facteur tissulaire] active alors le facteur IX. L'accumulation de facteur IX activé en présence de son cofacteur le facteur VIII activé, de phospholipides et d'ions calcium (complexe antihémophilique) permettra secondairement l'activation du facteur X en facteur X activé.
 Le déclenchement de la coagulation. Il est admis que l élément déclenchant de la coagulation in vivo est l expression à la surface des cellules (monocytes, cellules endothéliales voire fibroblastes) de facteur tissulaire. Le facteur tissulaire est un récepteur membranaire de très haute affinité pour le facteur VII. Il est normalement absent de la circulation sanguine mais est exprimé au niveau des cellules musculaires lisses de la paroi vasculaire et des fibroblastes de façon constitutive. Certains tissus sont très riches en facteur tissulaire : tissu cérébral par exemple. Ainsi, Lors d une brèche vasculaire ou lors de l expression pathologique du facteur tissulaire par certaines cellules sanguines, ce dernier fixe le facteur VII circulant qu’il active formant un complexe: [facteur VII activé - facteur tissulaire]. Il existe une toute petite quantité préalable de facteur VII déjà activé dans le plasma mais qui en l’absence de facteur tissulaire a très peu d’activité enzymatique. A partir de la formation du complexe, deux voies d activation sont possibles (Fig.4): Quand le facteur tissulaire est en excès, le complexe [facteur VII activé - facteur tissulaire]. active directement le facteur X. Cette voie peut cependant être rapidement inhibée par l’inhibiteur de la voie du facteur tissulaire, le TFPI. Quand le facteur tissulaire est en faible quantité (ou l inhibition par le TFPI prépondérante), le complexe [facteur VII activé - facteur tissulaire] active alors le facteur IX. L accumulation de facteur IX activé en présence de son cofacteur le facteur VIII activé, de phospholipides et d ions calcium (complexe antihémophilique) permettra secondairement l activation du facteur X en facteur X activé.")
22
La phase d’initiation Formation de la tenase extrinsèque: FVIIa - FT
La fixation du facteur VII au facteur tissulaire permet l’activation du facteur VII. Le facteur VII activé peut soit activer directement le facteur X (si le facteur tissulaire est en excès), soit activer le facteur IX qui en présence de facteur VIII activera le facteur X.
, soit activer le facteur IX qui en présence de facteur VIII activera le facteur X.")
23
Le FXa converti de petite quantité de prothrombine (FII) pour donner la thrombine (FIIa) en faible quantité incapable de transformer le Fibrinogène en Fibrine. - Cette quantité faible de FIIa initialement formée active le FV et le FVIII. Le complexe FT-FVIIa forme avec le FXa un complexe FVIIa- FT- FXa qui sera tout de suite inhibé par son inhibiteur naturel: le TFPI.

25
b - La phase d’amplification
Le FIIa initialement formée active le FVIII, le FV et le FXI et les plaquettes. - Le FIX peut être activé par deux mécanismes différents: * soit par le FXIa à la surface des plaquettes activées * soit par le complexe FT-FVIIa - L’activation du FVIII permet la formation à la surface de plaquette activée d’un complexe FVIIIa – FIXa appelé: Tenase intrinsèque.

26
La phase d’amplification

27
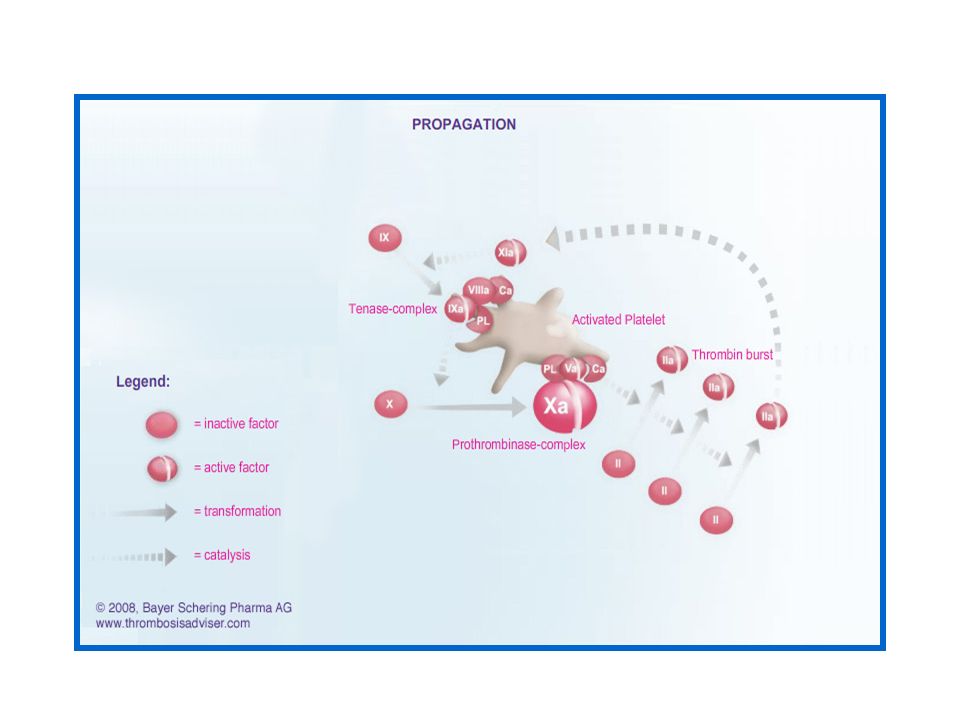
c - La phase de propagation et de stabilisation
Le complexe (FVIIIa–FIXa) permet une activation rapide du FX au niveau de la surface cellulaire des plaquettes activées. Le FXa en association avec le FVa constitue le Complexe Prothrombinase ( FVa, FXa, PL, Ca2+ ). Ce complexe Prothrombinase est responsable d’une génération abondante de thrombine .
 permet une activation rapide du. FX au niveau de la surface cellulaire des plaquettes. activées. Le FXa en association avec le FVa constitue le Complexe. Prothrombinase ( FVa, FXa, PL, Ca2+ ). Ce complexe Prothrombinase est responsable d’une. génération abondante de thrombine .")
28
La phase de propagation et de stabilisation

29
Sous l’action de la thrombine, le Fibrinogène va être
dégradé en monomère de Fibrine. Ces monomères de Fibrine vont se transformer en polymères de Fibrine instables encore solubles. Le FXIII activé par la Thrombine transforme ces polymères instables en polymères stables et insolubles.

31
Ces polymères insolubles forment un réseau qui va emprisonner des GR pour former le thrombus rouge.

32
La cascade de la coagulation
1-Initiation Brèche vasculaire 2-Amplification 3-Propagation Vue d‘ensemble de la cascade de la coagulation selon la nouvelle théorie avec avec ses 4 phases. Point de départ: lésion vasculaire Résultat: Transformation du Fg soluble en Fb insoluble. formation de thrombus rouge pour colmater la brêche 4-Stabilisation Thrombus rouge

33
Régulation de la coagulation in vivo
Pour éviter une activation diffuse et continue du processus de la coagulation, chaque facteur possède son inhibiteur spécifique. On connaît trois systèmes d’inhibiteurs principaux - Le système de l’Antithrombine (AT). - Le système Protéine C - Protéine S et Protéine Z ( PC-PS-PZ). - Le TFPI: Tissue Factor Pathway Inhibitor
. - Le système Protéine C - Protéine S et. Protéine Z ( PC-PS-PZ). - Le TFPI: Tissue Factor Pathway Inhibitor.")
34
Schéma général (2 voies)
FVIIa - FT - Ca2+ XIIa,XIa,PK,KHPM IXa,VIIIa,PL,Ca2+ TFPI IX PC-PS AT PZ X Xa, Va, PL, Ca2+ AT Dès qu'apparaissent des traces de thrombine, le processus de coagulation s'amplifie. La thrombine casse le fibrinogène en libérant 2 petits peptides : fibrinopeptide A et fibrinopeptide B. En perdant le fibrinopeptide A puis le fibrinopeptide B, le fibrinogène devient la fibrine. Spontanément, les monomères de fibrine peuvent se polymériser et former un premier réseau ou polymère soluble de fibrine. Ce polymère est instable. Il sera stabilisé par un dernier facteur, le facteur XIII (facteur stabilisant la fibrine: FSF). Le facteur XIII crée des liaisons covalentes solides entre ces monomères de fibrine. On a alors formation d'un réseau de fibrine qui emprisonne les globules rouges : le thrombus rouge définitif est ainsi formé (Fig 5). Prothrombine Thrombine Fibrinogène Fibrine
. Le facteur XIII crée des liaisons covalentes solides entre ces monomères de fibrine. On a alors formation d un réseau de fibrine qui emprisonne les globules rouges : le thrombus rouge définitif est ainsi formé (Fig 5). Prothrombine. Thrombine. Fibrinogène. Fibrine.")
35
Mécanisme de la coagulation in vivo
Mécanisme de la coagulation in vivo Le complexe FVII-FT initialise la génération du FXa et de FIIa en quantité suffisante pour activer le FV, le FVIII l’activation et l’agrégation locale des plaquettes. 2- Le complexe FVIIa- FT- FXa sera tout de suite inhibé par son inhibiteur naturel: le TFPI.

36
Mécanisme de la coagulation in vivo
Mécanisme de la coagulation in vivo 3- La quantité de FXa générée par le complexe FVII-FT n’est plus suffisante pour maintenir la coagulation. 4- La génération du FXa sera amplifiée par la voie du FIXa – FVIIIa pour assurer la coagulation.

37
EXPLORATION DE LA COAGULATION

38
Tests de 1ère intention Le Temps de Quick (TQ)
EXPLORATION DE LA COAGULATION Tests de 1ère intention Le Temps de Quick (TQ) Le Temps de Céphaline + Activateur (TCA) Fibrinogène (Fg) Temps de thrombine (TT)
 Le Temps de Céphaline + Activateur (TCA) Fibrinogène (Fg) Temps de thrombine (TT)")
39
Exploration de la coagulation: Tests globaux
FVIIa - FT - Ca2+ XIIa,XIa,PK,KHPM TCA TQ IX IXa,VIIIa,PL,Ca2 + TQ + TCA X Xa, Va, PL, Ca2+ Prothrombine Thrombine Dès qu'apparaissent des traces de thrombine, le processus de coagulation s'amplifie. La thrombine casse le fibrinogène en libérant 2 petits peptides : fibrinopeptide A et fibrinopeptide B. En perdant le fibrinopeptide A puis le fibrinopeptide B, le fibrinogène devient la fibrine. Spontanément, les monomères de fibrine peuvent se polymériser et former un premier réseau ou polymère soluble de fibrine. Ce polymère est instable. Il sera stabilisé par un dernier facteur, le facteur XIII (facteur stabilisant la fibrine: FSF). Le facteur XIII crée des liaisons covalentes solides entre ces monomères de fibrine. On a alors formation d'un réseau de fibrine qui emprisonne les globules rouges : le thrombus rouge définitif est ainsi formé (Fig 5). TQ + TCA Fg + TT Fibrinogène Fibrine
. Le facteur XIII crée des liaisons covalentes solides entre ces monomères de fibrine. On a alors formation d un réseau de fibrine qui emprisonne les globules rouges : le thrombus rouge définitif est ainsi formé (Fig 5). TQ + TCA. Fg + TT. Fibrinogène. Fibrine.")
40
Importance de l’étape pré analytique
Conditions de prélèvement Centrifugation Congélation et décongélation.

41
Explore les facteurs : VII, II, V, X, Fibrinogène. Allongement :
Temps de Quick (TQ) Explore les facteurs : VII, II, V, X, Fibrinogène. Allongement : AVK (INR), héparine ? Déficit quantitatif ou qualitatif en un ou plusieurs facteurs (allongement isolé déficit en facteur VII) Peu sensible aux A.C.C.
 Explore les facteurs : VII, II, V, X, Fibrinogène. Allongement : AVK (INR), héparine Déficit quantitatif ou qualitatif en un ou plusieurs facteurs (allongement isolé déficit en facteur VII) Peu sensible aux A.C.C.")
42
Expression du TQ - En % - Rapport = temps malade temps témoin - INR (au cours du traitement par AVK)
 .")
43
Temps de Céphaline + Activateur (TCA)
Explore les facteurs: - VIII, IX, XI et XII, PK, KHPM - Fibrinogène et le facteur II : moins sensible - Sensible aux A.C.C, et l’héparine Importance du choix du réactif

44
TCA : Valeurs Normales Varient en fonction du réactif et de l’instrumentation. Raccourcissement du TCA : Activation de la coagulation au moment du prélèvement (mauvaise qualité technique) Facteur VIII augmenté (Inflammation). Le TCA varie avec l’âge (plus allongé chez l’enfant).
 Facteur VIII augmenté (Inflammation). Le TCA varie avec l’âge (plus allongé chez l’enfant).")
45
Rapport = temps malade / temps témoin
Expression du TCA Rapport = temps malade / temps témoin Adulte < 1,22 Femme enceinte <1,20 Nouveau né < 1,70

46
TCA isolément allongé Inhibiteur de la coagulation Déficit en l’un des facteurs de la coagulation explorés par le TCA et non par le TQ. Distinction entre les 2 situations : Épreuve de correction

47
TCA : Épreuve de Correction
Choix du plasma témoin : - Pool de plasmas normaux congelés à -80°C (double centrifugation), ou plasma lyophilisé de commerce. - Éviter le choix « d’un plasma témoin du jour » Réaliser 3 TCA en parallèle : Temps malade, Temps témoin et Temps du mélange Malade + Témoin
, ou plasma lyophilisé de commerce. - Éviter le choix « d’un plasma témoin du jour » Réaliser 3 TCA en parallèle : Temps malade, Temps témoin et Temps du mélange Malade + Témoin.")
48
Épreuve de Correction Calculer l’Indice de Rosner: t(M+T) – tT x tM Indice < 12: Correction Le diagnostic de déficit(s) en facteur est(sont) évoqué(s). Qui porte nécessairement sur un ou plusieurs des facteurs suivants : VIII, IX, XI, XII, prékallicréine, kinininogène de haut poids moléculaire.
 en facteur est(sont) évoqué(s). Qui porte nécessairement sur un ou plusieurs des. facteurs suivants : VIII, IX, XI, XII, prékallicréine, kinininogène de haut poids moléculaire.")
49
Déficit en facteur(s) Déficit en :
- FVIII (hémophilie A), FIX (hémophilie B), FXI : manifestations hémorragiques: ++ Déficits en : - FXII, prékallicréine, KHPM : absence de manifestations hémorragiques.
, FIX (hémophilie B), FXI : manifestations hémorragiques: ++ Déficits en : - FXII, prékallicréine, KHPM : absence de manifestations. hémorragiques.")
50
DIAGNOSTIC D’UN ALLONGEMENT ISOLE DU TCA
Temps de Thrombine TT Normal TT Allongé TCA (T+M) Temps de Reptilase Nl Correction: DEFICIT Non correction: INHIBITEUR HEPARINE VIII IX XI Willebrand ACC LA XII, PK, KHPM
 Temps de Reptilase. Nl. Correction: DEFICIT. Non correction: INHIBITEUR. HEPARINE. VIII. IX. XI. Willebrand. ACC LA. XII, PK, KHPM.")
51
Allongement isolé du TQ
- Déficit isolé en FVII - Début de traitement par les AVK

52
Allongement simultané du TQ et TCA
Déficits en Facteurs I, II, V, ou X. Fibrinogène diminué: Défibrination(CIVD), exceptionnellement afibrinogénémie ou hypofibrinogénémie. Fibrinogène normal, TT allongé: Héparine, exceptionnellement dysfibrinogénémie Fibrinogène et TT normaux: - Déficits en II, V ou X: exceptionnels -Hypovitaminose K (V normal) - Insuffisance hépatocellulaire (V abaissé)
, exceptionnellement afibrinogénémie ou hypofibrinogénémie. Fibrinogène normal, TT allongé: Héparine, exceptionnellement dysfibrinogénémie. Fibrinogène et TT normaux: - Déficits en II, V ou X: exceptionnels. -Hypovitaminose K (V normal) - Insuffisance hépatocellulaire (V abaissé)")
53
EXPLORATION DE L’HEMOSTASE
Tests d’orientation Tests complémentaires Hémostase primaire Numération plaquettaire et Temps de saignement Dosage du F.VW, Agrégation plaquettaire Coagulation intrinsèque TCA Dosage des FVIII, FIX, FXI et FXII Coagulation extrinsèque TQ Dosage du FVII Voie commune TCA et TQ Dosage des FII, FV et FX Fibrinoformation TCA , TQ, Fibrinogène, Temps de Thrombine, Temps de Reptilase Dosage du FXIII,

54
Les maladies hémorragiques de la coagulation

55
I- Définition Hémorragie:
Perte de sang à partir d’une artère ou d’une veine. Elle peut être externe ou interne. Maladies hémorragiques: Ensemble de pathologies caractérisées par une tendance hémorragique de façon spontanée ou provoquée.

56
II- Caractères cliniques
1- Hématomes Collection de sang superficielle ou profonde qui se forme à la suite d’un choc et ne peut pas gagner l’extérieur de la peau. Elles peuvent être graves selon leur localisation (rétro-péritonéale, rétro-orbitale, psoas ….)
")
57
2- Hémarthroses Epanchement sanguin au niveau des articulations (genoux, coudes, chevilles).
.")
58
Les hématomes et les hémarthroses sont caractéristiques de
l’hémophilie A ou B.

59
3- Hémorragies internes
Le plus souvent graves: - Hématuries - hémorragies digestives - hémorragies cérébrales …….

60
L’HEMOPHILIE a- Définition Anomalie constitutionnelle de la coagulation qui relève d’une triple définition: - Cliniquement: elle s’exprime par un syndrome hémorragique caractéristique associant dans sa forme majeure: des hémarthroses et des hématomes profonds. - Génétiquement: L’anomalie se situe sur le chromosome X, et son expression est liée au sexe. - Biologiquement: Elle est due à l’absence ou à un défaut qualitatif du FVIII (hémophilie A) ou du FIX (hémophilie B)
 ou du FIX (hémophilie B).")
61
b- Génétique - Mode de transmission: Maladie récessive liée au chromosome X: Seul les garçons sont atteints alors que les filles sont conductrices et en général n’expriment pas la maladie.

62
Mode de transmission XHY (Hémophile) XX XY XY XHX XHX
 XX XY XY XHX XHX")
63
Mode de transmission XY XHX (conductrice ) XY XHY XHX XX
 XY XHY XHX XX")
64
c- Manifestations cliniques La sévérité de la maladie est directement liée aux degrés de déficit en FVIII ou en FIX. On distingue: l’hémophilie majeure, mineure et modérée. A taux égal, la sévérité est identique pour l’hémophilie A et B et, au sein d’une même famille, elle est identique d’un membre à l’autre.

65
Forme majeure Syndrome hémorragique qui apparait dans la petite enfance, le plus souvent lors de l’apprentissage de la marche. Deux types d’hémorragies dominent les manifestations cliniques de l’hémophilie majeure: les hémarthroses et les hématomes.

66
- Les hémarthroses Les articulations les plus touchées sont celles non ou peu protégées par la masse musculaire: genoux, coudes, chevilles, épaules, hanches ….

67
La gravité de ces hémarthroses tient à leur caractère récidivant au même endroit aboutissant progressivement à l’arthropathie hémophilique

68
- Les hématomes Collection de sang bien limitée superficielle ou profonde. Ces hématomes sont le plus souvent provoqués pouvant toucher tous les territoires mais ce sont surtout les hématomes profonds qui sont graves.

69
- Hématome du psoas (muscle au niveau des vertèbres lombaires) qui peuvent simuler une urgence chirurgicale (appendicite) - Hématome du cou avec risque d’asphyxie. - Hématome périorbitaire avec risque de cécité.

70
Formes modérées et mineures Tableau hémorragique souvent atypique et discret, généralement révélé lors d’une situation à risque hémorragique au cours, par exemple, d’une intervention chirurgicale ou d’un traumatisme.

71
d- Diagnostic biologique
d- Diagnostic biologique 1- Diagnostic positif - Bilan d’hémostase standard: permet de suspecter le DC. - TS et chiffre plaquettaire normaux - TQ, TT, Fg normaux - TCA allongé - Confirmation: dosage des facteurs VIII et IX 2- Précision de la sévérité de la maladie - Hémophilie majeure: FVIII ou FIX < 1% - Hémophilie modérée: 1% < FVIII ou FIX < 5% - Hémophilie mineure: 5%< FVIII ou FIX<30%

72
e - Traitement 1- Traitement préventif Repose sur la prise en charge globale de l’hémophile et de sa famille. - Le problème médical doit être régulièrement évalué par une équipe médicale pluridisciplinaire. - Chaque malade doit disposer d’une carte d’hémophile ( type de l’hémophilie, sévérité, règles de conduite générale…)
.")
73
2- Traitement substitutif Hémophilie A - Produits plasmatiques: Hémofil*, FVIII LFB* - Produits recombinants: Recombinate* Dose: Poids(Kg) X Augmentation souhaitée/ 2 1 unité FVIII/ Kg augmente le taux FVIII de 2%
 X Augmentation souhaitée/ 2 1 unité FVIII/ Kg augmente le taux FVIII de 2%")
74
Hémophilie B - Produits plasmatiques: Monime. , FIX LBR
Hémophilie B - Produits plasmatiques: Monime*, FIX LBR* - Produits recombinants: Benefix* Dose: Poids(Kg) X Augmentation souhaitée. 1 unité FIX/ Kg augmente le taux FIX de 1 à 1.5%
 X Augmentation souhaitée. 1 unité FIX/ Kg augmente le taux FIX de 1 à 1.5%")
75
LES DEFICITS CONGENITAUX EN FACTEURS DE LA COAGULATION
Les déficits en facteurs II, V, VII, X - Affections rares, dont le DC est évoqué soit devant un syndrome hémorragique (déficit sévère) soit fortuitement (déficit modéré). - Transmission selon le mode autosomal récessif - La sévérité des manifestations hémorragiques est liée au degré du déficit.
 soit fortuitement (déficit modéré). - Transmission selon le mode autosomal récessif. - La sévérité des manifestations hémorragiques est liée au degré du déficit.")
76
Déficit en FII: Ecchymoses, ménorragies, hémarthroses si déficit sévère
TQ et TCA allongé, FII abaissé Traitement: PPSB Déficit en FV: Hémorragies cutanéo-muqeuses, post-traumatiques ou chirurgicales. TQ et TCA allongé, FV abaissé Traitement: PFC

77
Déficit en FVII: Précoce et sévère, hémorragie du SNC, ménorragies,
hémarthroses TQ allongé, TCA normal, FVII abaissé Traitement: PPSB ou concentré de FVII Déficit en FX: Précoce (chute de cordon), hémorragies du SNC, chirurgicales, ménorragies. TQ et TCA allongés, FX abaissé Traitement: PPSB
, hémorragies du SNC, chirurgicales, ménorragies. TQ et TCA allongés, FX abaissé. Traitement: PPSB.")
78
Transmission autosomale dominante, particulièrement fréquent
Les déficits en facteurs contact Peu fréquents, voire exceptionnels. A l’exception du déficit en FXI, ils ne s’accompagnent d’aucun syndrome hémorragique, ni spontané, ni traumatique, ni en situation chirurgicale. Déficit en FXI Transmission autosomale dominante, particulièrement fréquent dans la population juive ashkénase. Hémorragies le plus souvent post-traumatiques ou post-chirurgicales, volontiers retardées et prolongées. TCA allongé, TQ normal FXI < 10% chez les homozygotes Traitement: PFC ou concentré de FXI purifié ( Hemoleven* )
")
79
Transmission autosomale récessive.
Déficit en FXII, PK, KHPM Ne sont plus considérés comme des facteurs de la coagulation selon la nouvelle théorie de la coagulation Transmission autosomale récessive. Aucune manifestation hémorragique, même en cas de déficit sévère. Au contraire, ce déficit expose aux thromboses. TCA allongé > à 100 sec chez l’homozygote, corrigé par incubation prolongée (15 min) du plasma avec le céphaline + activateur en cas de déficit en PK. Aucun traitement même en cas de déficit sévère
 du plasma avec le céphaline + activateur en cas de. déficit en PK. Aucun traitement même en cas de déficit sévère.")
80
Les déficits en facteurs de la fibrinoformation
Très rares, voire exceptionnels. Transmission autosomale récessive Afirinogénémie Transmission autosomale récessive Manifestations hémorragiques précoces et sévères, provoquées et prolongées: hémorragie à la chute de cordon, hémorragies du SNC et des muqueuses. TS souvent allongé et Fg indosable. Traitement: perfusion de Fg. Dysfirinogénémies Transmission autosomale dominante Manifestations hémorragiques totalement absentes, parfois discrètes voire même des manifestations thrombotiques. Allongement discret des TQ et TCA, Allongement important du TT/ T Reptilase

81
Déficit en FXIII Transmission autosomale récessive. Seuls les homozygotes sont symptomatiques : Hémorragies précoces à la chute de cordon surtout provoquées et retardées, hémorragies du SNC, Hémarthroses. Hémorragies postopératoires souvent associées à des troubles de cicatrisation Caillot soluble dans l’acide monochloroacétique 1%. Déficit en FXIII Traitement: perfusion de cc de Fg riches en FXIII.

Présentations similaires












>")







