Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Sclérose Latérale Amyotrophique : 1 Généralités
Dr Dupuy, Service de Neurologie CHU Amiens

2
Familiale dans 10% des cas Prévalence= 7 cas pour 100 000 hab
= Maladie de Charcot Maladie neurodégénérative incurable, appartient maladies du motoneurone Familiale dans 10% des cas Prévalence= 7 cas pour hab Incidence= 2 nouveaux cas pour hab. et par an Possiblement familiale : image donnée par les ascendants, pondération émotionnelle et affective, héritage des sentiments, de l ’histoire et des angoisses… Relativement fréquente à peine un peu moins fréquente que la SEP et les tumeurs cérébrales. Chiffres révisées à la haute en raison de l ’augmentation de l ’espérance de vie par contrôle des maladies cardio vasculaires et en raison de l ’augmentation de l ’espérance de vie des patients SLA Population encore jeune Pas de cause unique indentifiée nombreuses hypothèses sur des facteurs favorisants ou précipitant connaissances de plus en plus précises sur les mécanismes intimes ( au niveau chimique et cellulaire) de la dégénérescence : les essais médicamenteux et le RILUZOLE mais : un médicament comme cela a fait faire des bons en avant dans la prise en charge : plurisdciplinarité, solidarité, recrutement des malades, organisation, espoir.
 de la dégénérescence : les essais médicamenteux et le RILUZOLE. mais : un médicament comme cela a fait faire des bons en avant dans la prise en charge : plurisdciplinarité, solidarité, recrutement des malades, organisation, espoir.")
3
Physiopathologie et rappels
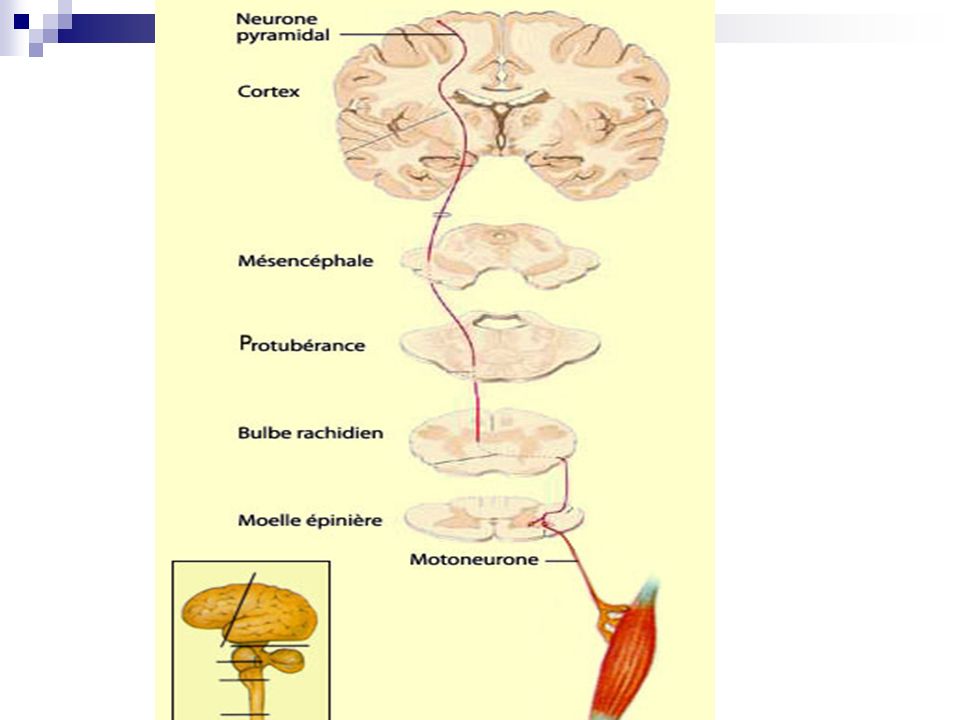
Dégénerescence progressive des motoneurones α de la corne antérieure de la moelle et du bulbe rachidien: syndrome neurogène et bulbaire + Dégénérescence des voies pyramidales: syndrome pyramidal et pseudobulbaire

5
Rappels Syndrome pyramidal: Syndrome pseudobulbaire:
paralysie spastique, contracture, ROT vifs, Babinski Syndrome pseudobulbaire: troubles de la mimique, rire et pleurer spasmodique, dysarthrie (voix nasonnée), troubles de la déglutition, paralysie nerfs crâniens (abolition R nauséeux et vélopalatin), réflexe palmomentonnier + et massétérin vif Phase chronique syndrome pyramidal Pas s associés syndrome pseudo bulbaire
, troubles de la déglutition, paralysie nerfs crâniens (abolition R nauséeux et vélopalatin), réflexe palmomentonnier + et massétérin vif. Phase chronique syndrome pyramidal. Pas s associés syndrome pseudo bulbaire.")
6
Rappels Syndrome neurogène périphérique: Syndrome bulbaire:
paralysie avec amyotrophie, fasciculations, ROT abolis ou diminués, troubles vaso moteurs Syndrome bulbaire: paralysie labio-glosso-pharyngée: dysarthrie, troubles de la déglutition (R nauséeux aboli et vélopalatin aboli que d’un côté) et amyotrophie linguale avec fasciculations
 et amyotrophie linguale avec fasciculations.")
7
10% des patients ont moins de 40 ans
âge de début moyen: 60 ans 10% des patients ont moins de 40 ans Diagnostic très difficile au début Délai diagnostique très fréquent (moy = 11 mois) médiane de survie= 36 mois mais extrêmes 6 mois-10 ans (10%) Annonce diagnostique difficile Cette rapidité évolutive complexifie beaucoup la prise en charge, nous y reviendrons tout le travail pour aller le moins mal possible , tout ce travail que font le patient, sa famille et les soignants demande du temps et du temps on en a pas beaucoup. médiane de survie à 71,5 mois si début avant 44 ans C ’est pourquoi on a été considérablement aidé par le Rilutek puis la VNI : on RALENTIT le processus , on gagne de la vie à un stade où elle est appréciée, tout en gagnant en qualité de survie, nous en reparlerons ! On ne se borne pas à une médiane de survie mais on laisse place à l’incertitude, à la singularité et on s’adapte aux demandes patient, selon entourage et capacités cognitives
 médiane de survie= 36 mois mais extrêmes 6 mois-10 ans (10%) Annonce diagnostique difficile. Cette rapidité évolutive complexifie beaucoup la prise en charge, nous y reviendrons. tout le travail pour aller le moins mal possible , tout ce travail que font le patient, sa famille et les soignants demande du temps et du temps on en a pas beaucoup. médiane de survie à 71,5 mois si début avant 44 ans. C ’est pourquoi on a été considérablement aidé par le Rilutek puis la VNI : on RALENTIT le processus , on gagne de la vie à un stade où elle est appréciée, tout en gagnant en qualité de survie, nous en reparlerons ! On ne se borne pas à une médiane de survie mais on laisse place à l’incertitude, à la singularité et on s’adapte aux demandes patient, selon entourage et capacités cognitives.")
8
Les maladies du motoneurone
Atteinte motoneurone central (MNC) = spasticité -paraplégies spastiques familiales -sclérose latérale primitive Atteinte motoneurone périphérique (MNP) = phénotype amyotrophie spinale -amyotrophies spinales progressives -syndrome post-polio Atteinte mixte = SLA Amyotrophies Spinales Progressives (ASP) et syndromes apparentés (Stark-Kaeser, O’Sullivan et McLoed, Maladie de Kennedy, Maladie d’Hyrayama) la distinction entre SLA et SLP peut être difficile (Le Forestier et al, 2001 ; Kuipers-Upmeijer et al, 2001). En faveur de la SLA on retiendra les données électromyographiques en faveur d’une atteinte du motoneurone périphérique. En faveur de la SLP, Kuipers-Upmeijer et coll. proposent de retenir l’allongement des temps de conduction centraux (Potentiels évoqués moteurs) et l’atrophie corticale focale du gyrus précentral en IRM. C’est dans ces formes cliniques que peuvent être découverts les rares syndromes paranéoplasiques de cancers solides qui ont pu mimer une SLA ou une SLP, plus souvent en rapport avec un cancer du sein (Forsyth et al, 1997). amyotrophie très focalisée à 1 membre ou segment de membre : syndrome d’Hirayama (anti GM1 ou trouble mécanique) syndrome post poliomyélitique ancienne polio aiguë dans la petite enfance stabilisation pendant au moins 10 ans dégénérescence motoneuronale des années plus tard ( + de 30 ans) sans inclusion ni signe inflammatoire révélée par une fatigue, une intolérance au froid (faiblesse musculaire et douleurs) défaillance des capacités de réinnervation ? évolution très lente “ bénigne ” les amyotrophies spinales de l’adulte : .la maladie de Kennedy Les amyotrophies spinales progressives SMN du Ch 5, Adulte type 4 : atrophie proximale, hypertrophie des mollets Ch 7 : Syndrome de O’Sullivan et Mc Leod (atrophie membres sup) Ch 12 : syndrome de Stark Kaeser, syndrome scapulo Jambier
 = spasticité. -paraplégies spastiques familiales. -sclérose latérale primitive. Atteinte motoneurone périphérique (MNP) = phénotype amyotrophie spinale. -amyotrophies spinales progressives. -syndrome post-polio. Atteinte mixte = SLA. Amyotrophies Spinales Progressives (ASP) et syndromes apparentés (Stark-Kaeser, O’Sullivan et McLoed, Maladie de Kennedy, Maladie d’Hyrayama) la distinction entre SLA et SLP peut être difficile (Le Forestier et al, 2001 ; Kuipers-Upmeijer et al, 2001). En faveur de la SLA on retiendra les données électromyographiques en faveur d’une atteinte du motoneurone périphérique. En faveur de la SLP, Kuipers-Upmeijer et coll. proposent de retenir l’allongement des temps de conduction centraux (Potentiels évoqués moteurs) et l’atrophie corticale focale du gyrus précentral en IRM. C’est dans ces formes cliniques que peuvent être découverts les rares syndromes paranéoplasiques de cancers solides qui ont pu mimer une SLA ou une SLP, plus souvent en rapport avec un cancer du sein (Forsyth et al, 1997). amyotrophie très focalisée à 1 membre ou segment de membre : syndrome d’Hirayama (anti GM1 ou trouble mécanique) syndrome post poliomyélitique. ancienne polio aiguë dans la petite enfance. stabilisation pendant au moins 10 ans. dégénérescence motoneuronale des années plus tard ( + de 30 ans) sans inclusion ni signe inflammatoire. révélée par une fatigue, une intolérance au froid (faiblesse musculaire et douleurs) défaillance des capacités de réinnervation évolution très lente bénigne les amyotrophies spinales de l’adulte : .la maladie de Kennedy. Les amyotrophies spinales progressives. SMN du Ch 5, Adulte type 4 : atrophie proximale, hypertrophie des mollets. Ch 7 : Syndrome de O’Sullivan et Mc Leod (atrophie membres sup) Ch 12 : syndrome de Stark Kaeser, syndrome scapulo Jambier.")
9
Signes cliniques au début
Forme à début spinal (une main amyotrophiée, un steppage, une fatigabilité à l’effort, des douleurs musculaires….) Forme à début bulbaire (un nasonnement, une hypersialorrhée, des fausses routes aux liquides, un défaut d’élocution….) Forme respiratoire (un sommeil perturbé, un essoufflement, une fatigue, une somnolence…) Forme avec troubles cognitifs (une modification du caractère…, rares cas de démence)
 Forme à début bulbaire (un nasonnement, une hypersialorrhée, des fausses routes aux liquides, un défaut d’élocution….) Forme respiratoire (un sommeil perturbé, un essoufflement, une fatigue, une somnolence…) Forme avec troubles cognitifs (une modification du caractère…, rares cas de démence)")
10
Les symptômes initiaux sont exclusivement moteurs, spinaux et/ou bulbaires
Déficits moteurs progressifs (au début, la maladie ne s’exprime qu’à un ou quelques groupes musculaires voisins) Troubles du tonus articulaire (instabilité par hypotonie ou au contraire raideur) Les déficits concernent les muscles spinaux (membres, axe) ou bulbaires
 Troubles du tonus articulaire (instabilité par hypotonie ou au contraire raideur) Les déficits concernent les muscles spinaux (membres, axe) ou bulbaires.")
11
Au niveau bulbaire troubles de l’élocution, la phonation, la déglutition, la mimique, le bavage.

12
Examens paracliniques
Confirmer le diagnostic: examens électrophysiologiques: Electromyogramme: syndrome de corne antérieur et Potentiels évoqués moteurs: atteinte de la voie pyramidale Eliminer diagnostics différentiels: - faire IRM bulbo-médullaire: éliminer compression cervico bulbaire - cancer, lymphome, saturnisme… très rare Bilan du retentissement (nutritionnel, respiratoire, locomoteur, psychologique)
")
13
Évolution progressive sans rémission
Handicap, chutes Troubles nutritionnels Troubles respiratoires, fausses routes, infections Troubles de la communication La vie est bouleversée et la qualité de vie très altérée Évolution très différente d ’un malade à l ’autre, différent de internethandicap : ∑ déficiences / environnement Sa rapidité d’aggravation Son retentissement psychologiques, social et familial

14
La vie est menacée par l’atteinte des muscles respiratoires et les difficultés à déglutir
Insuffisance respiratoire restrictive par atteinte du diaphragme puis des muscles accessoires inspirateurs et expirateurs. Fausses routes. Dénutrition. Évolution très différente d ’un malade à l ’autre

15
L’adaptation psychologique est difficile
négation « je vais bien », évitement « je vis un jour à la fois », pensée magique « il y a un nouveau traitement qui va arriver», refus « je ne veux pas en parler», ou « … » impossibilité de parler Les suicides sont exceptionnels C’est une maladie vécue par toutes les personnes du noyau familial et affectif… Évolution très différente d ’un malade à l ’autre

16
Presque toutes les possibilités de commande motrice de l’organisme sont à terme concernées
soit un état comparable au Locked-in syndrome (« le scaphandre et le papillon », J D Bauby) seule communication possible repose sur: vigilance, capacités cognitives et intégrité des muscles oculomoteurs (qui ne sont pas toujours respectés si l’évolution est prolongée) Tous les patients ne vivent pas ce stade : car décès par arrêt cardiaques (dysautonomie, commande centrale) ,en particulier chez les plus âgés et dans certaines formes cliniques de la maladie. Cependant c ’est le stade que risque de vivre les malades qui sont trachéotomisés pour ventilation mécanique (VI)
 seule communication possible repose sur: vigilance, capacités cognitives et intégrité des muscles oculomoteurs (qui ne sont pas toujours respectés si l’évolution est prolongée) Tous les patients ne vivent pas ce stade : car décès par arrêt cardiaques (dysautonomie, commande centrale) ,en particulier chez les plus âgés et dans certaines formes cliniques de la maladie. Cependant c ’est le stade que risque de vivre les malades qui sont trachéotomisés pour ventilation mécanique (VI)")
17
Les causes de décès A retenir: on ne meurt pas étouffé
Insuffisance respiratoire restrictive chronique terminale dans 60% des cas : Soit majoration progressive des symptômes jusqu’au décès par coma hypercapnique. Soit décompensation respiratoire (infection ou encombrement ou atélectasie ou œdème pulmonaire) : 35% des cas Troubles du rythme cardiaque dans 5% à 18 % des cas. La Détresse respiratoire aiguë (« l’ asphyxie ») est exceptionnelle (fausse route alimentaire, embolie pulmonaire massive). Les infections et leur gravité sont favorisées par la dénutrition ON NE MEURT PAS ETOUFFE mais la prise en charge de la respiration est au cœur de nos préoccupation pour gagner de la vie et améliorer le confort Facteurs pronostics font de cette maladie une maldie singulière âge au début des symptômes forme clinique sexe délai diagnostique atteinte respiratoire: survie par VNI état nutritionnel facteurs génétiques prise en charge (Traynor et al, 2003, JNNP) : survie
 : 35% des cas. Troubles du rythme cardiaque dans 5% à 18 % des cas. La Détresse respiratoire aiguë (« l’ asphyxie ») est exceptionnelle (fausse route alimentaire, embolie pulmonaire massive). Les infections et leur gravité sont favorisées par la dénutrition. ON NE MEURT PAS ETOUFFE. mais la prise en charge de la respiration est au cœur de nos préoccupation pour gagner de la vie et améliorer le confort. Facteurs pronostics font de cette maladie une maldie singulière. âge au début des symptômes. forme clinique. sexe. délai diagnostique. atteinte respiratoire: survie par VNI. état nutritionnel. facteurs génétiques. prise en charge (Traynor et al, 2003, JNNP) : survie.")
18
Prise en charge: objectifs
Améliorer la qualité de vie Augmenter la survie Améliorer la qualité de vie des « aidants naturels » Assurer une fin de vie de qualité Prévenir les deuils pathologiques Prévenir le burn-out des soignants LA PLURIDISCIPLINARITE est la réponse à la complexité de la prise en charge qui risquerait d ’épuiser un seul praticien ou un seul soignant, et sa technicité qui profite de plus en plus de tous les progrès Elle est, pour le moment une réponse de l ’hôpital, encore même des CHRU (notion de centre SLA) mais attention, elle n ’est réellement profitable que dans l ’échange, la synthèse et le dialogue pour une conclusion au patient qui résulte de ce travail d ’équipe et s ’en trouve bonifiée. Il faudrait aussi que les soignants de proximité se sentent inclus dans cette pluridisciplinarité mais ils ont des contraintes et la pluridciplinarité (interdisciplinarité ) ne résiste pas longtemps à l ’éloignement géographique et temporel.
 mais attention, elle n ’est réellement profitable que dans l ’échange, la synthèse et le dialogue pour une conclusion au patient qui résulte de ce travail d ’équipe et s ’en trouve bonifiée. Il faudrait aussi que les soignants de proximité se sentent inclus dans cette pluridisciplinarité mais ils ont des contraintes et la pluridciplinarité (interdisciplinarité ) ne résiste pas longtemps à l ’éloignement géographique et temporel.")
19
Les traitements Médicaments visant le contrôle de la maladie par un impact sur le motoneurone: traitement neurotrophique ou neuroprotecteur: Rilutek ( survie 3 mois par an) et vitamine E Aide psychologique et/ou cognitive. Techniques de lutte contre le handicap spinal: aides à la mobilité et suppléances fonctionnelles. Compensation des difficultés à s’alimenter. Compensation des difficultés à communiquer. Compensation des difficultés respiratoires. Traitements des inconforts et douleurs. Aide humaine et sociale (et le rôle des associations: ARS). Phase terminale et décès.
 et vitamine E. Aide psychologique et/ou cognitive. Techniques de lutte contre le handicap spinal: aides à la mobilité et suppléances fonctionnelles. Compensation des difficultés à s’alimenter. Compensation des difficultés à communiquer. Compensation des difficultés respiratoires. Traitements des inconforts et douleurs. Aide humaine et sociale (et le rôle des associations: ARS). Phase terminale et décès.")
20
Symptômes pénibles fatigabilité neuro-musculaire et générale
faiblesse motrice, paralysies et incapacités, dépendance perte des possibilités d’expression difficultés et inconfort posturaux douleurs, crampes, spasticité sialorrhée, altérations stomatologiques transit

21
Importance d’ informer de l’ARS: association
rire et pleurer spasmodiques inquiétudes quant à la fin de vie: comment va-t-on mourir ? souffrance « proximologique » SLA = proches Importance d’ informer de l’ARS: association

22
Ergothérapie/ Kiné économie neuro-musculaire
ergonomie et aménagement du logement (progressif, préventif et palliatif) réadaptation palliative continue -mobilité/gestualité -communication -activités quotidiennes -positionnements (st. assise, st. couchée) -transferts -interaction avec l’environnement
 réadaptation palliative continue. -mobilité/gestualité -communication -activités quotidiennes. -positionnements (st. assise, st. couchée) -transferts -interaction avec l’environnement.")
23
Mobilité, positionnements et transferts
-ATTELLES (AT) de mobilité -DEAMBULATION : cannes et déambulateurs (extérieur, intérieur) -FAUTEUIL ROULANT DE LOCATION FAUTEUIL ROULANT D’ACQUISITION (électrique ? - options - aménagements) -STATION ASSISE DE CONFORT positionnement (tronc, tête, MS, MI) alternance positions et appuis -STATION COUCHEE lit, matelas, surmatelas et installations - « calages » appareils de TRANFERTS adaptés
 de mobilité. -DEAMBULATION : cannes et déambulateurs (extérieur, intérieur) -FAUTEUIL ROULANT DE LOCATION FAUTEUIL ROULANT D’ACQUISITION. (électrique - options - aménagements) -STATION ASSISE DE CONFORT positionnement (tronc, tête, MS, MI) alternance positions et appuis. -STATION COUCHEE. lit, matelas, surmatelas et installations - « calages » appareils de TRANFERTS adaptés.")
24
AT de communication (+++) (en relation avec orthophoniste)
écrire: outil, support clavier ambulatoire ( orthèse) ordinateur : interface logiciel (clavier à l’écran) fonction souris ou défilement épeler par désignation visuelle sur tableau transparent oui/non sur questions alphabet énoncé N.B. : A.T. = essais coût / délais suivi évolutif parc de prêt (ARS) ?
 ordinateur : interface. logiciel (clavier à l’écran) fonction souris ou défilement. épeler par désignation visuelle sur tableau transparent. oui/non sur questions alphabet énoncé. N.B. : A.T. = essais coût / délais suivi évolutif parc de prêt (ARS)")
25
2. Sclérose Latérale Amyotrophique : prise en charge des complications nutritionnelles et respiratoires

26
Physiopathologie de la dénutrition
Troubles de déglutition Dépression Inhalation Carences d’apport Anorexie Amyotrophie Insuffisance respiratoire Dénutrition Immuno-dépression Modifications métaboliques Pneumopathies Inflammation SLA : conférence de consensus

27
Surveillance de l’état nutritionnel
Indice de masse corporel (IMC) anormal si < 18,5 kg/m²si moins de 75 ans < 21 kg/m²si plus de 75 ans amaigrissement/ poids de forme si 5% en un mois 10% en 6 mois
 anormal si. < 18,5 kg/m²si moins de 75 ans. < 21 kg/m²si plus de 75 ans. amaigrissement/ poids de forme si. 5% en un mois. 10% en 6 mois.")
28
Prise en charge des troubles nutritionnels
Se mettre dans de bonnes conditions (compagnie, temps, installation assise buste vertical/cou fléchi; bonne position de l’aidant) Se concentrer sur le repas (“penser sa déglutition” : Ø distraction ni conversation) Prendre de petites bouchées, 1 par 1 Fermer lèvres et mâchoires pour mastiquer ou déglutir Etre prêt à faire face à une situation d’urgence (manoeuvre d’Heimlich) Ne pas utiliser un liquide pour faire passer un solide
 Se concentrer sur le repas ( penser sa déglutition : Ø distraction ni conversation) Prendre de petites bouchées, 1 par 1. Fermer lèvres et mâchoires pour mastiquer ou déglutir. Etre prêt à faire face à une situation d’urgence (manoeuvre d’Heimlich) Ne pas utiliser un liquide pour faire passer un solide.")
29
Prise en charge des troubles nutritionnels
Adapter la texture de l’alimentation et hydratation Fractionnement de l’alimentation et enrichissement Eviter les aliments trop sucrés ou acides (car salivation) Poudre épaississante pour boissons Favoriser les boissons chaudes et surtout les froides ou pétillantes Eaux gélifiées (coûteuses/ pb acceptation) Compléments oraux (1 à 2 par jour)
 Poudre épaississante pour boissons. Favoriser les boissons chaudes et surtout les froides ou pétillantes. Eaux gélifiées (coûteuses/ pb acceptation) Compléments oraux (1 à 2 par jour)")
30
Indications de la suppléance nutritionnelle et choix de celle-ci
Compléments oraux dès que apports inférieurs aux besoins Nutrition entérale proposée si Fausses routes Durée excessive des repas Perte de poids Choix suppléance en fonction de l’état respiratoire : GPE: gastrostomie per endocopique +++ GPR: gastrostomie par voie radiologique Sonde naso gastrique

32
Surveillance respiratoire
Car pronostic et vitesse de dégradation Variabilité individuelle ++ Pour déterminer le moment optimal de prise en charge VNI: ventilation non invasive Désencombrement Et permettre de discuter et délibérer sur la prise en charge respiratoire terminale Selon un rythme de suivi trimestriel

33
Évaluation respiratoire
Clinique: Troubles du sommeil : dyssomnie, ronflement ou disparition récente du ronflement, céphalées matinales, somnolence diurne. Signes respiratoires discrets et tardifs chez des patients à mobilité réduite lors du décubitus : intolérance ou balancement thoraco- abdominal ou accélération de la fréquence respiratoire au repos ou à la parole : dyspnée, respiration «haute », tirage cervical, sueurs. Encombrement Epreuves fontionnelles respiratoires ( EFR) et évaluation nocturne
 et évaluation nocturne.")
34
Traitement des troubles respiratoires par Ventilation non invasive (VNI):
gain de survie de 15 mois en moyenne amélioration de la qualité de vie progrès techniques (masques, désencombrement) limites de la VNI= celles des masques formes bulbaires Sous VNI, les patients font moins souvent le choix d’une VI par trachéotomie.
 limites de la VNI= celles des masques formes bulbaires. Sous VNI, les patients font moins souvent le choix d’une VI par trachéotomie.")
35
Traitement des troubles respiratoires par Ventilation Invasive et trachéotomie: résultats.
Pour 70 % des malades,VI car atteinte respiratoire d’emblée sévère sur un diagnostic de SLA de moins de 1 an. Johnston, ALS/MND Milan 2003. Contexte de décompensation aiguë = pronostic immédiat médiocre: 50 % de décès à 1 mois (Cellerin et al, 1994) Survies prolongées ≥10 ans (Simon et al, 1999; Yoshida et al, 1992; Cazzolli et Oppenheimer, 1996) Situations de Locked-in syndrom (Kotchoubey et al 2003)
 Survies prolongées ≥10 ans. (Simon et al, 1999; Yoshida et al, 1992; Cazzolli et Oppenheimer, 1996) Situations de Locked-in syndrom. (Kotchoubey et al 2003)")
36
et donne une valeur aux directives anticipées.
La Loi relative au droit des malades et à la fin de la vie du 22 avril 2005 reconnaît un droit aux malades au moment particulier qu’est la fin de la vie qu’elle définie comme étant la « phase avancée ou terminale d’une affection grave et incurable, quelle qu’en soit la cause » : « les actes de soins ne doivent pas être poursuivis par une obstination déraisonnable » définie par des critères d’inutilité et de disproportion, offre la possibilité « de suspendre ou de ne pas entreprendre des soins n’ayant d’autre effet que le seul maintien artificiel de la vie », à la condition d’assurer des soins de confort tels que définis par l’article L du Code de la Santé Publique, et donne une valeur aux directives anticipées.

37
Les essentiels 1) Maladie neurodégénérative, maladie du motoneurone la plus fréquente 2) Dégénerescence progressive de des motoneurones α de la corne antérieure de la moelle et du bulbe rachidien: syndrome neurogène et bulbaire + 3) Dégénérescence des voies pyramidales: syndrome pyramidal et pseudobulbaire 4) Vers 60 ans en général, non génétique (10%), moyenne de survie de 36 mois 5) Forme spinale: paralysie motrice progressive 6) Forme bulbaire: trouble de la phonation, déglutition
 Dégénerescence progressive de des motoneurones α de la corne antérieure de la moelle et du bulbe rachidien: syndrome neurogène et bulbaire. + 3) Dégénérescence des voies pyramidales: syndrome pyramidal et pseudobulbaire. 4) Vers 60 ans en général, non génétique (10%), moyenne de survie de 36 mois. 5) Forme spinale: paralysie motrice progressive. 6) Forme bulbaire: trouble de la phonation, déglutition.")
38
Les essentiels 7)Maladie différente suivant chaque patient
8)Décès par insuffisance respiratoire chronique ou cardiaque, asphyxie rarrissime 9) Incurable mais prise en charge palliative dès le début, multidisciplinaire: neuro, rééeducateur, kiné, orthophoniste, ergothérapeute, diététicienne, pneumologue, psychologue, soins palliatifs 10) Questions éthiques de la suppléance des fonctions vitales, patient et qualité de vie du patient au centre de la réflexion
Décès par insuffisance respiratoire chronique ou cardiaque, asphyxie rarrissime. 9) Incurable mais prise en charge palliative dès le début, multidisciplinaire: neuro, rééeducateur, kiné, orthophoniste, ergothérapeute, diététicienne, pneumologue, psychologue, soins palliatifs. 10) Questions éthiques de la suppléance des fonctions vitales, patient et qualité de vie du patient au centre de la réflexion.")
Présentations similaires










>")









>")