Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Les affections mendéliennes
Modes de transmission autosomique dominant autosomique récessif récessif lié à l’X dominant lié à l’X mitochondrial Touchent ~ 1% de la population Fréquence très variable d’une maladie à l’autre Hémochromatose 2/1000 Mucoviscidose : 1/2500 Maladie de Wilson : 1/ Atteinte du SNC dans plus de 40%

2
Les affections mendéliennes
Nombre élevé de maladies différentes :

3
Les affections mendéliennes

4
Symboles généalogiques
sexes féminin 1 2 3 IV masculin indéterminé Jumeaux monozygotes Ordre de figuration Jumeaux dizygotes 3 2 3 2 3 garçons 6 2 garçons, 3 filles 2 filles ? 6 enfants Zygotisme incertain Sujets atteints Mariage Sujets décédés Proposant Grossesse en cours Femme conductrice d’une maladie liée à l’X Union illégitime puis mariage consanguin Individus hétérozygotes pour une maladie récessive Fausse couche Sujet adopté Couple sans descendants Sujets examinés sains

5
Exemple de maladies autosomiques dominantes
Hypercholestérolémie familiale (récepteur LDL cholestérol) Syndrome de Marfan (gène de la fibrilline) Achondroplasie (FGFR3) Neurofibromatose type I (NF1) Maladie de Huntington (HD, …) Maladie de Steinert (DMPK) Ostéogénèse imparfaite (collagène type I) Polykystose rénale (PKD1, PKD2) Porphyries Maladie de Charcot-Marie de type 1
 Syndrome de Marfan (gène de la fibrilline) Achondroplasie (FGFR3) Neurofibromatose type I (NF1) Maladie de Huntington (HD, …) Maladie de Steinert (DMPK) Ostéogénèse imparfaite (collagène type I) Polykystose rénale (PKD1, PKD2) Porphyries. Maladie de Charcot-Marie de type 1.")
6
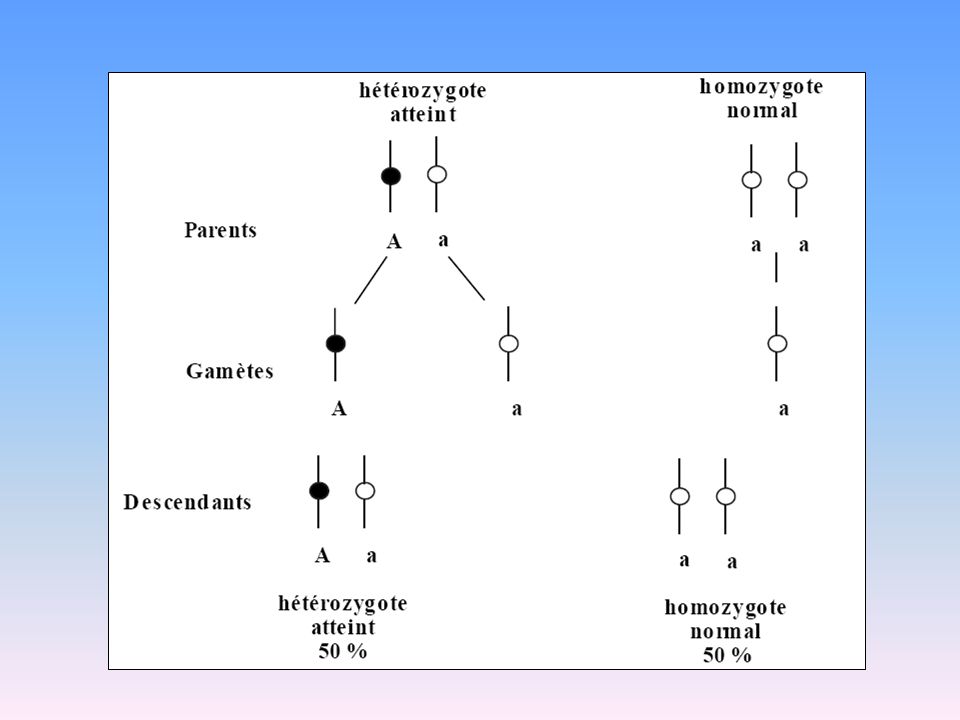
Transmission Autosomique Dominante
Dominant : caractère qui exprime son phénotype lorsqu’il est présent à l’état hétérozygote aussi bien qu’homozygote

8
Lois de la transmission AD
Un sujet malade a un parent malade (sauf mutation de novo) La transmission est verticale sans saut de génération : la maladie est transmise directement du père ou de la mère à son enfant La proportion de sujets malades est identiques dans les deux sexes La descendance de sujets sains est indemne Un sujet malade (hétérozygote) et un sujet normal auront une descendance ~ 50% d’enfants malades Un sujet homozygote ne peut provenir que de deux parents atteints Aa x aa Aa aa a A Aa x Aa Aa AA aa aA a A
 La transmission est verticale sans saut de génération : la maladie est transmise directement du père ou de la mère à son enfant. La proportion de sujets malades est identiques dans les deux sexes. La descendance de sujets sains est indemne. Un sujet malade (hétérozygote) et un sujet normal auront une descendance ~ 50% d’enfants malades. Un sujet homozygote ne peut provenir que de deux parents atteints. Aa x aa. Aa. aa. a. A. Aa x Aa. Aa. AA. aa. aA. a. A.")
9
Anomalies cardiovasculaires
Notion de Pléiotropie Une mutation unique peut provoquer des anomalies de plusieurs organes ou tissus, parfois sans lien apparent ex :Syndrome de Marfan du à des mutations du gène de la fibrilline (protéine de la matrice extracellulaire) Anomalies oculaires Anomalies squelettiques Anomalies cardiovasculaires
 Anomalies oculaires. Anomalies squelettiques. Anomalies cardiovasculaires.")
10
Pléiotropie : maladie de Wilson
Excrétion anormale du cuivre qui s’accumule dans les tissus

11
Notion de pénétrance Nombre d’individus phénotypiquement atteints
Nombre d’individus génotypiquement atteints Complète dans certaines maladies : - Syndrome de Marfan - Polypose rectolique - Sphérocytose… à condition de rechercher précisément le phénotype Incomplète dans d’autres maladies - Neurofibromatose de type 1, - Polydactylie… possibilité de sauts de génération Dépendante de l’âge : ex maladie de Huntington

12
Pénétrance incomplète
Exemple d’arbre Individus porteurs asymptomatiques Pénétrance = 6/12 = 50 %

13
Pénétrance dépendante de l’âge
Exemple de la maladie de Huntington Pénétrance (%) 30 40 50 60 70 Age (années)
 Age (années)")
14
Expressivité variable
Quantitative Ex : sévérité variable de la neurofibromatose de Recklinghausen " tâches de café au lait" Illustration originale de Von Recklinghausen en 1882

15
Expressivité variable
2. Qualitative Tâches de café au lait > 90% Aspect en peau de chagrin de la région axillaire > 90% Neurofibromatomes > 90% Nodules de l’iris > 90% Leucémies ou cancers fréquentes Retard mental % Macrocranie % Petite taille % Tumeurs du SNC <10%

16
Expressivité variable
3. Selon le sexe Ex : calvitie Trait mendélien autosomique dominant chez l’homme récessif chez la femme influencé par les hormones (mécanisme biochimique complexe) Sexe Calvitie Normal Homme BB, Bb bb Femme BB Bb, bb
 Sexe. Calvitie Normal. Homme. BB, Bb. bb. Femme BB. Bb, bb.")
17
Expressivité variable Gènes liès au sexe situés sur le chromosome X
3. Selon le sexe, lié à l’X Ex : Daltonisme, Hémophilie Gènes liès au sexe situés sur le chromosome X + = Vision Normale (Dominant) o = Daltonisme (Récessif) Sexe Daltonisme Vision Normale Homme XoY X+Y Femme XoXo X+X+ X+Xo
 o = Daltonisme (Récessif) Sexe. Daltonisme. Vision Normale Homme. XoY. X+Y. Femme. XoXo. X+X+ X+Xo.")
18
Expressivité variable
4. Selon le sexe du parent atteint (empreinte parentale) Dystrophie myotonique de Steinert Transmission maternelle des formes néonatales - Début âge adulte : myopathie, myotonie, calvitie, cataracte, hypogonadisme, troubles cardiaques - Forme congénitale : détresse respiratoire à la naissance, diplégie faciale, hypotonie et développement moteur retardé, retard mental, difficultés d’alimentation) Maladie de Huntington Transmission paternelle des formes juvéniles Délétion 15q11-q13 Paternelle : Syndrome de Prader-Willi Maternelle : Syndrome d’Angelman
 Dystrophie myotonique de Steinert. Transmission maternelle des formes néonatales. - Début âge adulte : myopathie, myotonie, calvitie, cataracte, hypogonadisme, troubles cardiaques. - Forme congénitale : détresse respiratoire à la naissance, diplégie faciale, hypotonie et développement moteur retardé, retard mental, difficultés d’alimentation) Maladie de Huntington. Transmission paternelle des formes juvéniles. Délétion 15q11-q13. Paternelle : Syndrome de Prader-Willi. Maternelle : Syndrome d’Angelman.")
19
Empreinte parentale Un gène est soumis à empreinte lorsque l'expression de ce gène dépend de son origine parentale (maternelle ou paternelle). Un gène peut être soumis à empreinte seulement dans un tissu particulier (par exemple uniquement dans le placenta) ou à un moment particulier (par exemple au cours du dévelopement embryonnaire). Les gènes soumis à empreinte sont le plus souvent regroupés dans des domaines chromatiniens contrôlés par un centre d'inactivation. On connaît actuellement chez l'homme plus de 30 gènes soumis à empreinte parentale, et on estime qu'il en existe probablement dix fois plus. Il existe, chez un individu diploïde, une disomie uniparentale (DUP) pour un chromosome ou un segment de chromosome lorsque les deux exemplaires de ce matériel ont été hérités d'un seul et même parent.
 ou à un moment particulier (par exemple au cours du dévelopement embryonnaire). Les gènes soumis à empreinte sont le plus souvent regroupés dans des domaines chromatiniens contrôlés par un centre d inactivation. On connaît actuellement chez l homme plus de 30 gènes soumis à empreinte parentale, et on estime qu il en existe probablement dix fois plus. Il existe, chez un individu diploïde, une disomie uniparentale (DUP) pour un chromosome ou un segment de chromosome lorsque les deux exemplaires de ce matériel ont été hérités d un seul et même parent.")
20
Région 15q11-q13 Syndrome d’Angelman Syndrome de Prader-Willi
1/20000 Syndrome de Prader-Willi 1/10000 Déficit mental sévère Langage absent Aspect joyeux, rires immotivés Ataxie, motricité saccadée Épilepsie Retard psychomoteur Retard de langage et d’apprentissage Comportement alimentaire impulsif Hypotonie Hypogonadisme Cause : perte de fonction du gène UBE3A (expression maternelle) Del 15q11 maternelle (70%) disomie uniparentale paternelle (2%) Mutations dans UBE3A (20%) Mutations du centre d’empreinte (<5%) Causes : perte de fonction des gènes PWS (expression paternelle) Del 15q11 paternelle (70%) disomie uniparentale maternelle (25%) Mutations du centre d’empreinte (<5%)
 Del 15q11 maternelle (70%) disomie uniparentale paternelle (2%) Mutations dans UBE3A (20%) Mutations du centre d’empreinte (<5%) Causes : perte de fonction des gènes PWS (expression paternelle) Del 15q11 paternelle (70%) disomie uniparentale maternelle (25%) Mutations du centre d’empreinte (<5%)")
21
Région de délétion-duplication 15q11-q13
~4.5-5 Mb Paternal expression domain Maternal expression domain ZNF127 UBE3A ATP10C GABRB3 GABRA5 GABRG3 HERC2 APBA2 NDN SNRPN OCA2 IPW 13 12 11.2 11.1 14 15 21.1 21.2 21.3 22.1 22.2 22.3 23 24 25 26.1 26.2 26.3 Chromosome 15

22
Exemple de transmission
Famille avec une disruption du centre d’empreinte responsable d’un Syndrome d’Angelman Buiting, Am J Hum Genet, 2001

23
Expressivité variable
5. Influence du milieu Hypercholestérolémie familiale : maladie AD due à une anomalie du récepteur aux LDL (low density proteins) Hétérozygotes (>1/1000) Augmentation d’un facteur 2 du cholestérol dans les LDL Xanthomes tendineux Risque élevé de décès par infarctus Prévention des complications cardiaques par régime et traitement hypocholestérolémiant Homozygotes (1/106) Manifestations + sévères avec décès avant 30 ans (infarctus du myocarde) Amélioration partielle par un traitement préventif
 Hétérozygotes (>1/1000) Augmentation d’un facteur 2 du cholestérol dans les LDL. Xanthomes tendineux. Risque élevé de décès par infarctus. Prévention des complications cardiaques par régime et traitement hypocholestérolémiant. Homozygotes (1/106) Manifestations + sévères avec décès avant 30 ans (infarctus du myocarde) Amélioration partielle par un traitement préventif.")
24
Expressivité variable
6. Influence d’autres gènes = épistasie Effet génétique résultant de l’interaction entre 2 gènes non homologues pour l’expression d’un caractère. Cet effet peut être additif ou de dominance Un allèle à un locus peut masquer ou modifier le caractère d’un allèle à un autre locus. Ex : Drépanocytose : Sexe : les femmes ont plus de HbS que les hommes Certaines populations (Sénégal) ont moins de [HbS] intracellulaire que d’autres (Benin, Cameroun) formes moins sévères de la maladie Cet effet est du à un polymorphisme (AT)x(T)y situé à 500 pb en amont du gène de la b-globine qui lie plus ou moins fortement la protéine BP1 et influence ainsi le niveau d’expression du gène.
 ont moins de [HbS] intracellulaire que d’autres (Benin, Cameroun) formes moins sévères de la maladie. Cet effet est du à un polymorphisme (AT)x(T)y situé à 500 pb en amont du gène de la b-globine qui lie plus ou moins fortement la protéine BP1 et influence ainsi le niveau d’expression du gène.")
25
Exemple d’épistasie récessive : le rare groupe sanguin humain "bombay"
Substance H Polysaccharide A (Enzyme d’ajout A) IAiH genotype A phenotype Polysaccharide B (Enzyme d’ajout B) IBiH genotype B phenotype iiH genotype O phenotype hh genotype Bombay phenotype Équivalent au phénotype O IAIBH genotype AB phenotype
 IAiH genotype. A phenotype. Polysaccharide B (Enzyme d’ajout B) IBiH genotype. B phenotype. iiH genotype. O phenotype. hh genotype. Bombay phenotype. Équivalent au phénotype O. IAIBH genotype. AB phenotype.")
26
Mutations de novo +/+ m/+ Ex de maladies où la proportion de néomutations est élevée : Achondroplasie (80%) Neurofibromatose de type 1 (50%) Maladie de Marfan (50%)
 Maladie de Marfan (50%)")
27
Mosaïques germinales Mosaïque : présence de deux populations de cellules, l’une étant porteuse d’une mutation, l’autre non +/+ m/+ Individu portant une mutation à l’état mosaïque Ex de maladies où un mosaïcisme germinal a été décrit : Ostéogénèse imparfaite Neurofibromatose 1 Myopathie de Duchenne

28
Taux de mutation A l’état d’équilibre, la fréquence d’une maladie dominante dans une population est stable car le nombre de porteurs de la mutation qui disparaissent en raison du désavantage sélectif est contre-balancé par ceux qui apparaissent par néomutation. Conséquence : dans une maladie grave qui altère la capacité à se reproduire, le taux de mutation est élevé dans une maladie grave qui ne modifie pas cette capacité, le taux de mutation est faible

29
Taux de mutation Maladie Taux de mutation / 106 gamète
Maladie de Huntington Syndrome de Marfan Achondroplasie Neurofibromatose <1 5 10 100 Le taux de mutation : n’est pas équivalent dans les 2 sexes augmente avec l’âge paternel dans certaines maladies (Syndrome de Marfan, achondroplasie)
")
30
Taux de mutation (1-f) µ M = 2µ + n (1-f) 1 M = M = 3 3 EX :
La proportion de cas dus à une mutation récente peut être calculée grâce à la formule de Haldane : M = proportion de cas dus à une mutation récente f = fertilité des garçons atteints µ = taux de mutation des gamètes femelles n = taux de mutation des gamètes mâles (1-f) µ M = 2µ + n (1-f) Si µ = n (taux de mutation identique dans les 2 sexes) 1 M = M = Si f = 0 3 3 EX : Maladie Myopathie de Duchenne Hémophilie A Maladie de Lesch-Nyhan Fertilité des garçons % de néomutations (M) Fréquence du gène (p) Taux de mutation (M x p) 0,8 1 / 3 ~ 7% 1 / 3 500 1 / 1 / ~ 1 / 1 / 1 / Taux de mutation plus élevé dans les gamètes males pour lesch-Nyhan. Fertilité nulle. Hémophilie A : mutations plus fréquentes dans les gamètes males. 3.1 times more often in males
 µ. M = 2µ + n. (1-f) Si µ = n (taux de mutation identique dans les 2 sexes) 1. M = M = Si f = EX : Maladie. Myopathie de Duchenne. Hémophilie A. Maladie de Lesch-Nyhan. Fertilité des garçons. % de néomutations (M) Fréquence du gène (p) Taux de mutation. (M x p) 0,8. 1 / 3. ~ 7% 1 / / / ~ 1 / / / Taux de mutation plus élevé dans les gamètes males pour lesch-Nyhan. Fertilité nulle. Hémophilie A : mutations plus fréquentes dans les gamètes males. 3.1 times more often in males.")
31
Effet fondateur Mutation survenue dans le passé chez un membre d’un groupe (fondateur) et transmise au sein de la même population Conséquence : Toutes les personnes atteintes dans la population concernées sont porteuse de la même mutation La fréquence de la maladie peut être élevée dans la population concernée Exemple : Trois couples d’anglais arrivés en Nouvelles Angleterre en 1630 sont à l’origine de plus de 1000 cas de maladie de Huntington tracés sur 12 générations

32
Effet fondateur (suite)
Population 1 Gènes Mutation Population 2 Population 3 Population 4

33
Transmission Autosomique Récessive
Récessif : caractère qui exprime son phénotype seulement lorsqu’il est présent à l’état homozygote On distingue : Homozygotes vrais porteurs de gènes identiques par transmission mendéliennes Hétérozygotes composites porteurs de gènes iso-actifs (mutations différentes d’un même gène entraînant le même phénotype Plus de 200 maladies autosomiques récessives résultent d’un déficit enzymatique caractérisé.

34
Exemple de maladies autosomiques récessives
Albinisme (plusieurs gènes) Mucoviscidose (CFTR) Drépanocytose (gène b globine) Thalassémies (gènes a ou b globines) Hémochromatose (HFE) Maladie de Tay-Sachs (TSD=HexA) Maladie de Gaucher (glucocérébrosidase) Maladie de Wilson (ATP7B) Amyotrophie spinale progressive (SMN1, SMN2) Ataxie de Friedreich (FRDA1 = gène de la frataxine) La plupart des maladies métaboliques dues à des anomalies enzymatiques, ex :phénylcétonurie (PAH) * *
 Mucoviscidose (CFTR) Drépanocytose (gène b globine) Thalassémies (gènes a ou b globines) Hémochromatose (HFE) Maladie de Tay-Sachs (TSD=HexA) Maladie de Gaucher (glucocérébrosidase) Maladie de Wilson (ATP7B) Amyotrophie spinale progressive (SMN1, SMN2) Ataxie de Friedreich (FRDA1 = gène de la frataxine) La plupart des maladies métaboliques dues à des anomalies enzymatiques, ex :phénylcétonurie (PAH) * *")
35
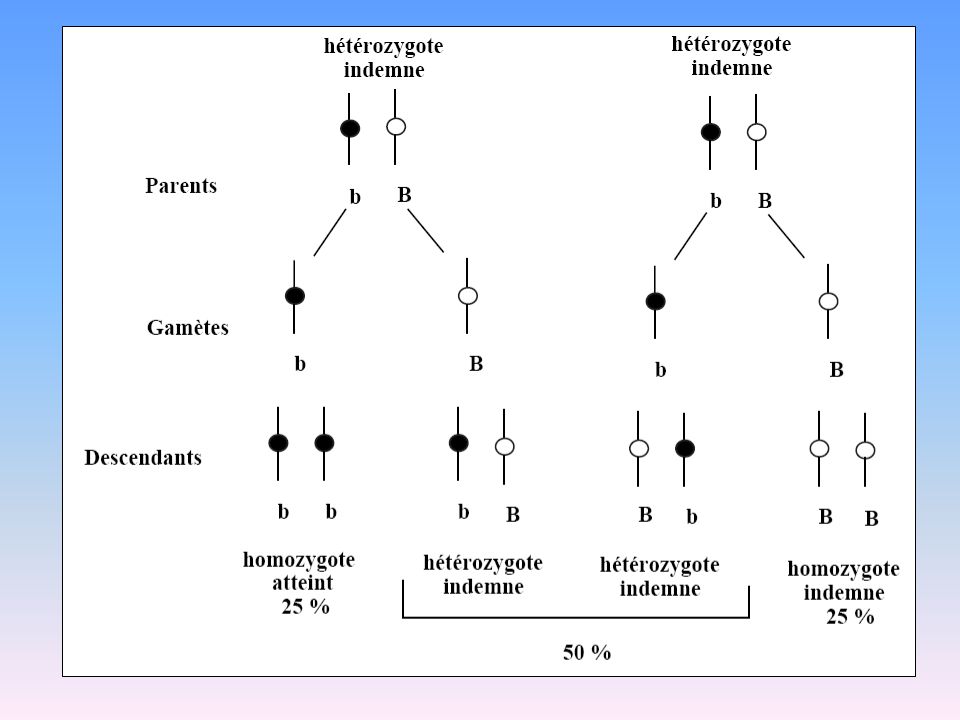
Transmission Autosomique Récessive

37
Lois de la transmission AR
L’union de deux hétérozygotes entraîne la naissance d’un sujet atteint pour 4 naissances La transmission est horizontale (n’atteint qu’une génération) les sujets atteints ont des parents normaux (hétérozygotes) La maladie s’exprime dans les deux sexes La proportion d’unions consanguines est élevée dans l’ascendance des sujets atteints (dans ces familles, le gène anormal à l’état homozygote chez les atteints provient de l’ancêtre commun) Aa x Aa a A A Aa AA a aa aA
 les sujets atteints ont des parents normaux (hétérozygotes) La maladie s’exprime dans les deux sexes. La proportion d’unions consanguines est élevée dans l’ascendance des sujets atteints. (dans ces familles, le gène anormal à l’état homozygote chez les atteints provient de l’ancêtre commun) Aa x Aa. a. A. A. Aa. AA. a. aa. aA.")
38
Lois de la transmission AR
5. L’union d’un homozygote atteint avec un homozygote sain donne naissance à des enfants tous hétérozygotes 6. L’union de deux sujets atteints entraîne la naissance d’enfants tous atteints AA x aa a a A Aa aA A Aa aA aa x aa a a a aa aa a aa aa

39
Le gène de la rousseur Transmission autosomique récessive pour la prédisposition à être roux Pénétrance incomplète Gène récepteur de la mélanocortine de type1 (MC1R) Dus à des variants de différents types de ce gène dont la fonction de signalisation est altérée Pénétrance dépend du variant : 3 variants de 0.79 Autre allèles pénétrance faible : 0.10 Hétérozygote : cheveux blonds
 Dus à des variants de différents types de ce gène dont la fonction de signalisation est altérée. Pénétrance dépend du variant : 3 variants de Autre allèles pénétrance faible : Hétérozygote : cheveux blonds.")
40
Teinte des cheveux en fonction des variants du gène MC1R chez des sujets indépendants

41
Loi de Hardy-Weinberg Établit une correspondance entre la fréquence des allèles et celle du génotype Fondée sur le fait que la probabilité de survenue de 2 évènements indépendants est le produit des probabilités de chaque événement. Soient 2 allèles : A de fréquence p a de fréquence q La distribution des génotypes dans une population est exprimée par : (p+q)2 = 1 = p2 +q2 + 2 pq Cette loi est vérifiée dans une population panmictique, sans mutation, sans migration q p q q2 qp p pq p2
2 = 1 = p2 +q2 + 2 pq. Cette loi est vérifiée dans une population panmictique, sans mutation, sans migration. q. p. q. q2. qp. p. pq. p2.")
42
Application de la loi de Hardy-Weinberg Calcul de la fréquence des hétérozygotes
Pour une maladie autosomique récessive dont la fréquence est de 1 / q2 = 1 / q = 1 / 100 d’où p = car p + q = 1 Fréquence des hétérozygotes = 2 p q = 2 x 99 / 100 x 1 / ~ 1 / 50 il y a donc 200 fois plus d’hétérozygotes porteurs du gène anormal que d’homozygotes atteints ~ 200 50 99 100

43
Nombre de porteurs / sujet atteint
Application de la loi de Hardy-Weinberg Calcul de la fréquence des hétérozygotes Exemples: Maladie Mucoviscidose Albinisme Maladie de Wilson Fréquence du gène des sujets atteints des porteurs sains Nombre de porteurs / sujet atteint 1 / 50 1 / 200 1 / 400 1 / 2 500 1 / 1 / 100 400 800 1 / 25 1 / 100 Conséquences La suppression des homozygotes attents affecte peu la fréquence du gène dans la population car la plupart des gènes mutés sont portés par des hétérozygotes sains

44
Consanguinité Consanguin : issu de l’union de sujets apparentés
Apparentés : sujets avec au moins un ancêtre commun Coefficient de consanguinité d’un individu : probabilité pour que 2 gènes en un locus soient identiques Risque : avoir des enfants atteints d’une maladie autosomique récessive Causes : - la plupart des gènes responsables d’affections autosomiques récessives sont portés par des hétérozygotes sains - 2 apparentés ont plus de chance que 2 individus non apparentés d’être porteurs de même allèle récessif

45
Coefficients de consanguinité
des enfants Type de mariage Père-fille / mère-fils Frère/Sœur Demi-frère/demi-sœur Oncle-nièce/tante-neveu Cousins germains Cousins issus de germains Cousins issus issus de germains 1/4 1/8 1/16 1/64 1/256 La proportion d’enfants atteints nés d’unions consanguines est plus élevée dans les maladies récessives dont le gène est rare (maladie de Wilson) que dans celles dont le gène est fréquent (mucoviscidose)
 que dans celles dont le gène est fréquent (mucoviscidose)")
46
Distribution des maladies autosomiques récessives selon les groupes de population
Syndrome néphrotique congénital aspartylglucosaminurie Mucoviscidose Phenylcétonurie Amish : Hypoplasie des cartilages et cheveux Juifs ashkénazes : Maladie de Tay-Sachs Maladie de Gaucher Dysautonomie familiale b-thalassémies Anémie falciforme (drépanocytose)
")
47
Exemple de la mucoviscidose

48
Transmission Récessive liée à l’X
Trait déterminé par des gènes du chr X qui se manifeste à l’état homozygote ou hémizygote Au moins 1/2000 enfants souffre d’une affection liée au chr X Ex d’affections récessives liées au chr X : Myopathies de Duchenne et Becker (gène de la dystrophine) Hémophilies A et B (gène de la facteurs VIII et IX de la coagulation) Syndrome de l’X fragile (FMR1) Maladie de Lesh-Nyhan (HPRT1) Choroïdérémie (CHM) Déficit en G6PD (gène de la G6PD) Adrénoleucodystrophie (ABCD1) Dystrophie musculaire d’Emery-Dreyfuss (Emerin, LMNA) Daltonisme (OPN1MW, OPN1LW) Diabète insipide néphrogénique (gène du récepteur de l’AVP V2)
 Hémophilies A et B (gène de la facteurs VIII et IX de la coagulation) Syndrome de l’X fragile (FMR1) Maladie de Lesh-Nyhan (HPRT1) Choroïdérémie (CHM) Déficit en G6PD (gène de la G6PD) Adrénoleucodystrophie (ABCD1) Dystrophie musculaire d’Emery-Dreyfuss (Emerin, LMNA) Daltonisme (OPN1MW, OPN1LW) Diabète insipide néphrogénique (gène du récepteur de l’AVP V2)")
49
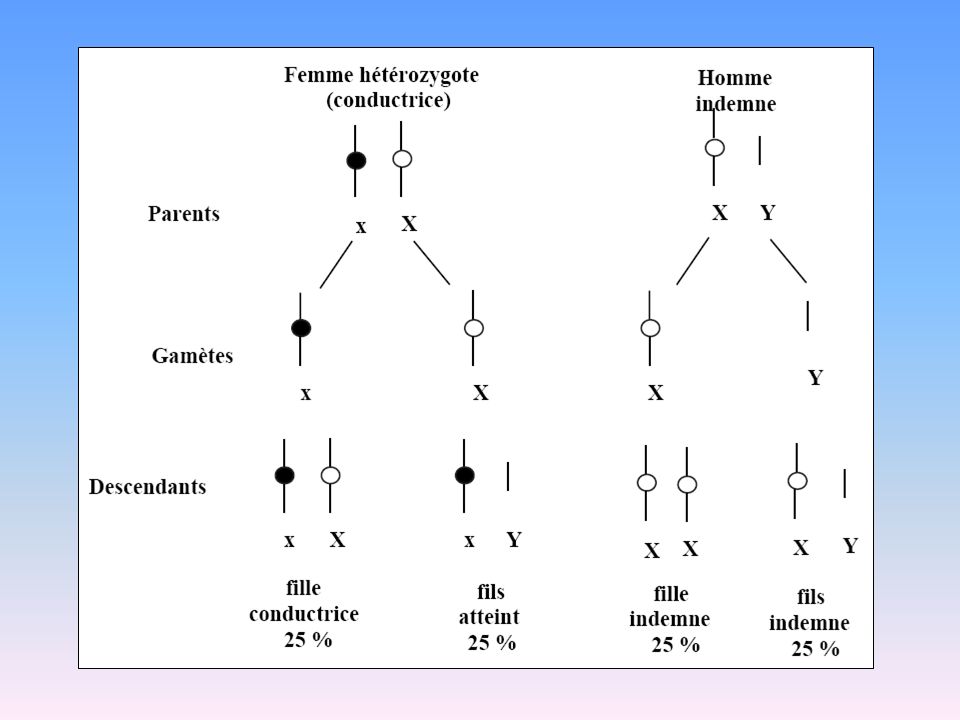
Transmission Récessive liée à l’X

51
Lois de la transmission récessive liée à l’X
Femme conductrice avec un homme sain 50% garçons atteints XaY 50% garçons sains XY 50% filles conductrices XaX 50% filles non conductrices XX XaX x XY X Y Xa XaX XaY X XX XY Les garçons atteints ont des mères conductrices (sauf en cas de néomutation) 2. Femme saine avec un homme atteint XaY x XX X X 100% garçons sains XY 100% des filles conductrices XaX Xa XaX XaX Y XY XY Pas de transmission père-fils
 2. Femme saine avec un homme atteint. XaY x XX. X. X. 100% garçons sains XY. 100% des filles conductrices XaX. Xa. XaX. XaX. Y. XY. XY. Pas de transmission père-fils.")
52
Lois de la transmission récessive liée à l’X
3. Femme conductrice avec un homme atteint (très rare) 50% garçons atteints XaY 50% garçons sains XY 50% filles atteintes XaXa 50% filles conductrices XaX XaX x XaY Xa Y Xa XaXa XaY X XaX XY
 50% garçons atteints XaY. 50% garçons sains XY. 50% filles atteintes XaXa. 50% filles conductrices XaX. XaX x XaY. Xa. Y. Xa. XaXa. XaY. X. XaX. XY.")
53
Transmission récessive liée au chromosome X (suite)
Atteinte beaucoup plus fréquente des hommes Si fréquence allèle Xa = q → fréquence des homes atteints = q (XaY) → fréquence des femmes atteintes = q2 (XaXa) Exemple : daltonisme q = 1/50 → fréquence des hommes atteints = 1/50 → fréquence des femmes atteintes = 1/2500 Situations particulières Femmes atteintes dans 3 situations: - Homozygotie (XaXa) - Syndrome de Turner (XaO), testicule féminisant (XaY) - Translocation Xa ; autosome (inactivation du X normal)
 → fréquence des femmes atteintes = q2 (XaXa) Exemple : daltonisme q = 1/50. → fréquence des hommes atteints = 1/50. → fréquence des femmes atteintes = 1/2500. Situations particulières. Femmes atteintes dans 3 situations: - Homozygotie (XaXa) - Syndrome de Turner (XaO), testicule féminisant (XaY) - Translocation Xa ; autosome (inactivation du X normal)")
54
Corpuscule de Barr (chr X inactivé)
Inactivation d’un chromosome X chez la femme : phénomène de lyonisation Lyonisation (phénomène découvert par Mary Lyon) : Inactivation de la majeure partie d’un des 2 chromosomes X de la femme - précoce (embryon) - aléatoire - indépendante d’une cellule à l’autre - définitive (le même chromosome X reste inactivé dans les cellules dérivant d’une cellule où il a été initialement inactivé) Embryon Corpuscule de Barr (chr X inactivé) Zygote X
 : Inactivation de la majeure partie d’un des 2 chromosomes X de la femme - précoce (embryon) - aléatoire. - indépendante d’une cellule à l’autre. - définitive (le même chromosome X reste inactivé dans les cellules dérivant d’une cellule où il a été initialement inactivé) Embryon. Corpuscule de Barr (chr X inactivé) Zygote. X.")
55
Inactivation d’un chromosome X
- Mécanismes : Processus d’inactivation initialisé au niveau d’un centre de contrôle sur le bras long du chr X; l’extrémité du bras court n’est pas concernée. Mécanisme moléculaire fait intervenir la méthylation des cytosines au niveau de séquences CpG - Conséquences : Chaque femme constitue une mosaïque somatique (Proportion variable de cellules dans lesquelles l’un ou l’autre des 2 chr X est inactivé) Compensation de l ’effet de dosage génique Hommes 1 chrX activé / Femmes 1 chrX activé + 1 chrX inactivé Une femme conductrice peut manifester des symptômes d’une maladie récessive liée à l’X Ex : près de 10% des femmes conductrices pour la myopathie de Duchenne présentent des manifestations cliniques (hypertrophie des mollets, faiblesse musculaire)
 Compensation de l ’effet de dosage génique. Hommes 1 chrX activé / Femmes 1 chrX activé + 1 chrX inactivé. Une femme conductrice peut manifester des symptômes d’une maladie récessive liée à l’X. Ex : près de 10% des femmes conductrices pour la myopathie de Duchenne présentent des manifestations cliniques (hypertrophie des mollets, faiblesse musculaire)")
56
Transmission Dominante liée à l’X
Trait porté par des gènes du chr X qui se manifeste aussi bien chez les hommes hémizygotes que chez les femmes hétérozygotes Très rare Rachitisme résistant à la vitamine D (gène récepteur de la vitD) Maladie de Charcot-Marie-Tooth dominante liée à l’X (Cx32) Syndrome Oro-facio-digital (OFD1) Déficit en Ornithine transcarbamylase (OTC) Parfois létal chez le garçon Incontinentia pigmenti
 Maladie de Charcot-Marie-Tooth dominante liée à l’X (Cx32) Syndrome Oro-facio-digital (OFD1) Déficit en Ornithine transcarbamylase (OTC) Parfois létal chez le garçon. Incontinentia pigmenti.")
57
Transmission Dominante liée à l’X

58
Lois de la transmission dominante liée à l’X
Femme atteinte avec un homme sain 50% garçons atteints XaY 50% garçons sains XY 50% filles atteintes XaX 50% filles saines XX XaX x XY X Y Xa XaX XaY X XX XY 50% des enfants atteints indépendamment de leur sexe 2. Femme saine avec un homme atteint XaY x XX X X 100% garçons sains XY 100% des filles atteintes XaX Xa XaX XaX Y XY XY Pas de transmission père-fils

59
Notion d’hétérogénéité génétique et allélique
Un même phénotype peut résulter de mutations de gènes différents situés à des loci distincts SCA1 SCA2 SCA3 SCA4 SCA6 SCA19/ SCA22 SCA26 SCA23 SCA25 SCA16 SCA8 DRPLA SCA12 SCA10 SCA11 SCA13 SCA14 SCA15 SCA27 SCA17 SCA18 SCA5/SCA20 SCA21 SCA7 Ex : loci responsables d’ataxies cérebelleuses AD

60
Notion d’hétérogénéité génétique et allélique
Ex : loci responsables de maladie de Charcot-Marie-Tooth Gènes Locus

61
Notion d’hétérogénéité génétique et allèlique
Hétérogénéité allélique Différentes altérations d’un même gène peuvent être resposables d’un même phénotype Ex : Mucoviscidose (AR) : plus de 350 mutations connues du gène CFTR Myopathie de Duchenne (lié à l’X) : plus de 150 mutations connues du gène DMD
 : plus de 350 mutations connues du gène CFTR. Myopathie de Duchenne (lié à l’X) : plus de 150 mutations connues du gène DMD.")
62
Particularités des mutations alléliques d’un gène
Un ou plusieurs phénotypes Un gène / un phénotype : mucoviscidose CFTR DMD Myopathie de Duchenne

63
Particularités des mutations alléliques d’un gène
Un ou plusieurs phénotypes Diversité phénotypique : un gène / plusieurs maladies Dystrophie musculaire d’Emery-Dreyfuss AD Progeria (maladie de Hutchinson-Gilford) Gène LMNA (lamines A/C) Maladie de Charcot-Marie-Tooth Lipodystrophie partielle
 Gène LMNA. (lamines A/C) Maladie de. Charcot-Marie-Tooth. Lipodystrophie partielle.")
64
Particularités des mutations alléliques d’un gène
2. Transmission récessive ou dominante Selon le type de mutation d’un même gène, l’affection sera transmise selon le mode dominant ou récessif Maladie Gène Transmission Rétinite pigmentaire Myotonie congénitale Thalassémie b Rhodopsine (RHO) Canal chlore CLCN1 b-globine (HBB) AD et AR
 Canal chlore CLCN1. b-globine (HBB) AD et AR.")
65
Mutations dynamiques (ou instables)
(CGG)n Normaux : 5-50, prémutés : , mutés : >230 FRAXA FRA11B FRAXE Inactivation du gène ex intr AUG UAA ex intr AUG UAA (CAG)n Normaux : 10-35, mutés : >35 Huntington SCA1,2,3… DRPLA Expansion toxique d’un domaine polyglutamine Normaux : 5-40, prémutés : 50-80, mutés : >80 (CUG)n ex intr AUG UAA Steinert (Gène DMPK) Interaction toxique avec des protéines de liaison aux messagers (GAA)n Normaux : 6-30, mutés : Ataxie de Friedreich (Gène FRDA1) Blocage de la transcription ex intr AUG UAA
n. Normaux : 5-50, prémutés : , mutés : >230. FRAXA. FRA11B. FRAXE. Inactivation du. gène. ex. intr. AUG. UAA. ex. intr. AUG. UAA. (CAG)n. Normaux : 10-35, mutés : >35. Huntington. SCA1,2,3… DRPLA. Expansion toxique d’un domaine polyglutamine. Normaux : 5-40, prémutés : 50-80, mutés : >80. (CUG)n. ex. intr. AUG. UAA. Steinert. (Gène. DMPK) Interaction toxique avec des protéines de liaison aux messagers. (GAA)n. Normaux : 6-30, mutés : Ataxie de Friedreich. (Gène FRDA1) Blocage. de la transcription. ex. intr. AUG. UAA.")
66
Phénomène d’anticipation
Ex : maladie de Huntington 40/17 18/20 15/20 44/20 42/20 17/18 44/15 60/17 57/18

67
Pathologie mitochondriale
1. Le génome mitochondrial Petite taille : pb 13 gènes (protéines de la chaîne respiratoire) ARN ribosomiques 12S et 16S 22 ARNt Présent en multiples copies 10 copies / mitochondrie 500 copies / cellule
 ARN ribosomiques 12S et 16S. 22 ARNt. Présent en multiples copies. 10 copies / mitochondrie. 500 copies / cellule.")
68
Pathologie mitochondriale
Transmission Hérédité maternelle Transmission des mitochondries par les ovocytes transmission mère-enfants (pas de transmission père-enfants)
")
69
Pathologie mitochondriale
3. Affections connues Au moins 12 connues dont : Atrophie optique de Leber Syndrome de Leigh Myopathie mitochondriale 4. Pénétrance incomplète et expressivité variable Hétéroplasmie Proportion variable d’ADN mitochondiral normal ou pathologique selon les cellules résultant des divisions cellulaires et de la prolifération des mitochondries Expression de la maladie au delà d’un seuil du rapport avec une sévérité croissante proportionnellement à ce rapport ADN muté ADN normal

70
Mutations pathologiques dans l’ARNt LEU (UUR)
PEM Progressive encephalopathy DMDF Diabetes Mellitus + DeaFness MM Mitochondrial Myopathy MELAS Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes MERRF Myoclonic Epilepsy and Ragged Red Muscle Fibers MMC Maternal Myopathy and Cardiomyopathy

71
Hérédité cytoplasmique I
- ADN mitochondrial : bp. Sans intron. - Gènes : 2 gènes d’ARN ribosomique, 22 gènes d’ARNt, 13 gènes codant des gènes de la chaîne respiratoire (codée aussi par le génome nucléaire). 1 seul transcrit, clivé ensuite - 2 à 10 copies d’ADNmt par mito. Nombreuses mito/cellule Génétique mitochondriale : Division des mitochondries indépendante de la division cellulaire Réplication de l’ADNmt au cours des phases S et G2 Ségrégation mitotique au hasard dans les cellules filles Hétéroplasmie : présence de molécules mutées et normales dans la même cellule, dans la même mito Homoplasmie : présence uniquement de molécules du même type
. 1 seul transcrit, clivé ensuite. - 2 à 10 copies d’ADNmt par mito. Nombreuses mito/cellule. Génétique mitochondriale : Division des mitochondries indépendante de la division cellulaire. Réplication de l’ADNmt au cours des phases S et G2. Ségrégation mitotique au hasard dans les cellules filles. Hétéroplasmie : présence de molécules mutées et normales dans la même cellule, dans la même mito. Homoplasmie : présence uniquement de molécules du même type.")
72
Génétique mitochondriale II
Hérédité cytoplasmique : maternelle. Les mères transmettent leur ADNmt à tous leurs enfants Transmission complexe : les enfants ne sont pas tous atteints, dépend de la proportion de molécules mutées. Maladies mitochondriales : touchent tous les organes -Accumulation d’équivalents réduits (NADH, FADH) -Mesure de la consommation d’oxygène par mito fraîches Mitochondries accumulées en périphérie des muscles Délétions hétéroplasmiques de grande taille (>1,5 kb), mutations ponctuelles
 -Mesure de la consommation d’oxygène par mito fraîches. Mitochondries accumulées en périphérie des muscles. Délétions hétéroplasmiques de grande taille (>1,5 kb), mutations ponctuelles.")
73
Définitions Génotype : constitution génétique d’un individu Phénotype : caractère observable de la constitution du génome d’un individu. Expression du génotype Locus : position de séquences d’ADN sur un chromosome (gènes ou fragments anonymes) Allèles : versions alternatives d’un gène. Par extension, variation de l’ADN en un locus ( polymorphisme) Homozygote : individu porteur d’allèles identiques à un locus donné (A/A) Hétérozygote : individu porteur d’allèles différents à un locus donné (A/a)
 Allèles : versions alternatives d’un gène. Par extension, variation de l’ADN en un locus ( polymorphisme) Homozygote : individu porteur d’allèles identiques à un locus donné (A/A) Hétérozygote : individu porteur d’allèles différents à un locus donné (A/a)")
Présentations similaires





>")
 Pr E. Tournier-Lasserve>")



 Pr E. Tournier-Lasserve>")


>")





