Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Médicaments psychotropes
La classification de Delay, Denicker

2
Classes Exemples Psycholeptiques ou sédatifs -Agissant sur la vigilance ou Nooleptiques · Hypnotiques Benzodiazépines -Agissant sur l'humeur ou Thymoleptiques · Neuroleptiques Phénothiazines Butyrophénones Benzamides · Anxiolytiques Thymorégulateurs ou normothymiques Sels de lithium Carbamazépine Psychoanaleptiques ou stimulants: - Stimulants de l'humeur ou Thymoanaleptiques: · Antidépresseurs IRS et IRSNA Tri et quadricycliques - Stimulants de la vigilance Nooanaleptiques Amphétamine et dérivés - Autres stimulants Xanthines Acide ascorbique Psychodysleptiques ou perturbateurs: Hallucinogènes ou onirogènes LSD Mescaline

3
ANXIOLYTIQUES 1-Les Benzodiazépines Actions pharmacologiques
Les principales actions des BDZ sont: 1. une réduction de l’anxiété et de l’agressivité Suppression des réponses émotionnelles psychiques comme somatiques (variations de la tension artérielle et du rythme cardiaque) 2. une sédation et une induction du sommeil (hypnotique) Elle explique les manifestations latérales de somnolence, d’apathie, de ralentissement des réflexes 3. une réduction du tonus musculaire (myorelaxante) et de la coordination Les BDZ diminuent le tonus des fibres striées par action centrale.
 2. une sédation et une induction du sommeil (hypnotique) Elle explique les manifestations latérales de somnolence, d’apathie, de ralentissement des réflexes. 3. une réduction du tonus musculaire (myorelaxante) et de la coordination. Les BDZ diminuent le tonus des fibres striées par action centrale.")
4
4. un effet anticonvulsivant
Tout déficit central de la transmission GABAergique se révèle épileptogène. Les substances facilitant cette médiation ont donc des propriétés anticonvulsivantes. 5. un effet amnésiant L’amnésie est antérograde et lacunaire. Elle ne porterait pas sur les événements antérieurs à la prise ni sur le moment de la prise et les minutes suivant la prise. Elle est de survenue aiguë et régresse spontanément en quelques heures.

5
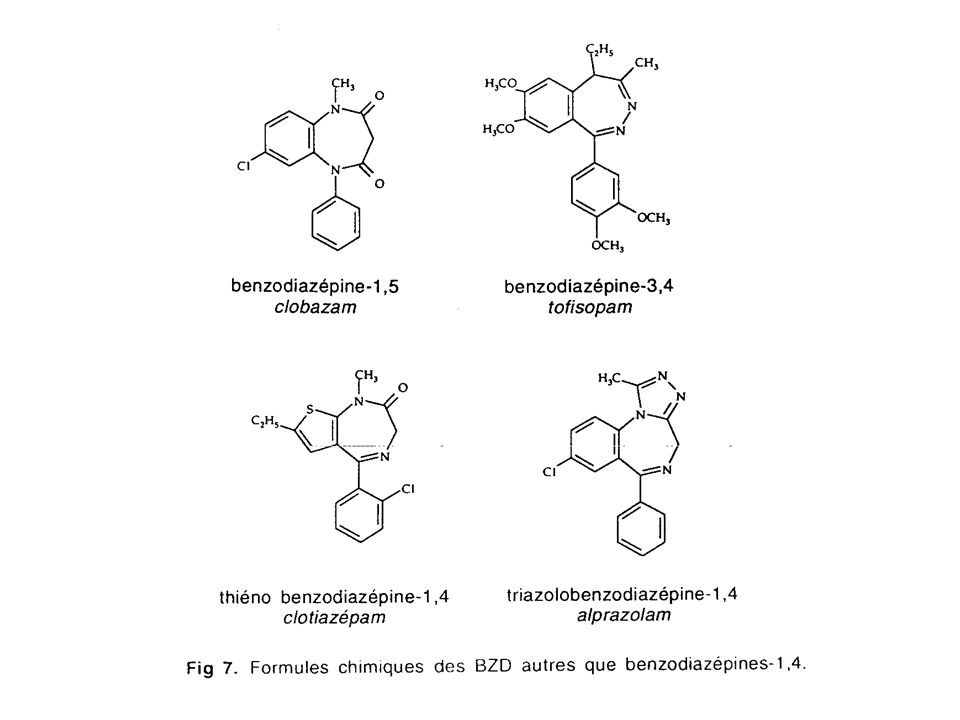
Structure des benzodiazépines

7
LE RÉCEPTEUR GABAA ET LES RÉCEPTEURS AUX BENZODIAZÉPINES
Mécanisme d’action LE RÉCEPTEUR GABAA ET LES RÉCEPTEURS AUX BENZODIAZÉPINES Le GABA est le premier acide aminé dont le rôle dans la neurotransmission fut reconnu La fonction dominante de l'acide gamma-aminobutyrique est son activité inhibitrice sur les neurones du SNC. Le système nerveux présente une concentration de GABA de 200 à 1000 fois supérieure à celle des autres neurotransmetteurs (acétylcholine, sérotonine...) : Dans la moelle : la substance grise, au niveau de la corne antérieure et en particulier dans la substance gélatineuse de Rolando, contient des interneurones GABAergiques. Cette localisation expliquerait l'activité myorelaxante des benzodiazépines.
 : Dans la moelle : la substance grise, au niveau de la corne antérieure et en particulier dans la substance gélatineuse de Rolando, contient des interneurones GABAergiques. Cette localisation expliquerait l activité myorelaxante des benzodiazépines.")
8
Dans le cervelet : Le GABA est le neuromédiateur d'interneurones inhibiteurs tels que les cellules en panier de Golgi et les cellules en étoile. Ces neurones envoient des efférences vers les cellules et les fibres excitatrices dont les neuromédiateurs sont les acides aspartique et glutamique. De même, les cellules de Purkinje GABAergiques sont les seuls neurones à envoyer des efférences hors du cervelet vers les noyaux profonds. Dans le système extrapyramidal : il existe également dans le striatum des interneurones courts régulateurs locaux et une voie GABAergique striato-nigrée freinant l'activité des voies dopaminergiques. Enfin, dans le cortex cérébral : il existe de très nombreux circuits interneuronaux GABAergiques régularisant l'excitabilité corticale.

9
Une telle fonction physiologique explique d'une part l’essor actuel en thérapeutique des molécules GABAmimétiques dans le traitement de certaines formes d'épilepsie et d'autre part l'activité antiépileptique des benzodiazépines.

10
Synthèse Le précurseur du GABA est la glutamine. La glutamine est synthétisée dans les cellules gliales à partir du glutamate (acide aminé excitateur) recapté de la fente synaptique. Cette réaction est catalysée par la glutamine synthétase, enzyme exclusivement localisée dans les cellules gliales. La glutamine est ensuite captée par les terminaisons axonales et transformée en glutamate par la glutaminase, enzyme mitochondriale. Le glutamate provient également de la transamination de l’alpha-cétoglutarate, produit de dégradation du glucose par le cycle de Krebs (CK). Le GABA est synthétisé par décarboxylation du glutamate grâce à une enzyme, la GAD (1 - Glutamic Acid Decarboxylase), présente dans la fraction cytosolique des terminaisons axonales GABAergiques. Cette enzyme a pour cofacteur le pyridoxal phosphate (PLP - ou vitamine B6). Ainsi, elle est inhibée par tous les agents qui la dissocient de son cofacteur, le PLP, auquel elle est faiblement liée.
 recapté de la fente synaptique. Cette réaction est catalysée par la glutamine synthétase, enzyme exclusivement localisée dans les cellules gliales. La glutamine est ensuite captée par les terminaisons axonales et transformée en glutamate par la glutaminase, enzyme mitochondriale. Le glutamate provient également de la transamination de l’alpha-cétoglutarate, produit de dégradation du glucose par le cycle de Krebs (CK). Le GABA est synthétisé par décarboxylation du glutamate grâce à une enzyme, la GAD (1 - Glutamic Acid Decarboxylase), présente dans la fraction cytosolique des terminaisons axonales GABAergiques. Cette enzyme a pour cofacteur le pyridoxal phosphate (PLP - ou vitamine B6). Ainsi, elle est inhibée par tous les agents qui la dissocient de son cofacteur, le PLP, auquel elle est faiblement liée.")
11
Le GABA GABA GLUTAMIC ACID DECARBOXYLASE GABA TRANSAMINASE

12
Une complexité supplémentaire a été introduite dans les schémas de la biosynthèse du GABA par la découverte de deux isoformes de la GAD. Ces deux isoformes, nommées GAD 65 et GAD 67, sont issues de deux gènes différents. Leurs masses moléculaires sont très proches et leurs séquences en acides aminés montrent qu’elles ont le même site actif. Cependant, elles diffèrent par leur localisation cellulaire et leur mode de fonctionnement vis à vis de leur cofacteur, le PLP : 1. La GAD 65 a une localisation préférentiellement axonale et n’est pas saturée en PLP. Son activité peut être augmentée par augmentation du taux de PLP dans les terminaisons; Ce mécanisme permettrait la production accrue et rapide de GABA. 2. La GAD 67, plutôt somato-dendritique, est saturée in vivo par le PLP, ce qui sous-tend probablement une production et une libération tonique de GABA par les neurones qui l’expriment. Ces deux mécanismes suggèrent l’existence d’une modulation très fine du métabolisme du GABA.

13
Recapture Après libération dans la fente synaptique, le GABA est capté par des transporteurs sélectifs (2) dans les neurones GABA et les cellules gliales. Ce sont des protéines membranaires à 12 segments transmembranaires hydrophobes. Le transport du GABA est dépendant des ions sodium (Na+) et chlore (Cl-). L’ion Cl- se fixe sur un site proche du site de fixation du GABA et augmente l’affinité du transporteur pour son substrat. Le gradient Na+ est nécessaire au transport du GABA dans la cellule, 2 ions Na+ étant transportés dans la cellule avec le GABA (symport 2 Na+ / 1 GABA). L’inversion du gradient Na+ provoque une libération de GABA à partir des neurones ou des cellules gliales. A l’heure actuelle, la biologie moléculaire a révélé l’existence de 4 types de transporteurs au GABA (GAT-1, GAT-2, GAT-3 et GAT-4) et leur complexité pharmacologique :
 dans les neurones GABA et les cellules gliales. Ce sont des protéines membranaires à 12 segments transmembranaires hydrophobes. Le transport du GABA est dépendant des ions sodium (Na+) et chlore (Cl-). L’ion Cl- se fixe sur un site proche du site de fixation du GABA et augmente l’affinité du transporteur pour son substrat. Le gradient Na+ est nécessaire au transport du GABA dans la cellule, 2 ions Na+ étant transportés dans la cellule avec le GABA (symport 2 Na+ / 1 GABA). L’inversion du gradient Na+ provoque une libération de GABA à partir des neurones ou des cellules gliales. A l’heure actuelle, la biologie moléculaire a révélé l’existence de 4 types de transporteurs au GABA (GAT-1, GAT-2, GAT-3 et GAT-4) et leur complexité pharmacologique :")
14
Le GABA GABA GLUTAMIC ACID DECARBOXYLASE GABA TRANSAMINASE

15
Le GAT-1 est exprimé dans le système nerveux et plus spécifiquement dans les neurones.
Le GAT-2 est localisé dans le cerveau, les reins et le foie et serait donc un transporteur glial. Le GAT-3 s’exprime dans le foie et le rein chez l’animal adulte. Il a donc toutes les caractéristiques d’un transporteur glial. Il s’exprime, cependant, très transitoirement dans le cerveau au cours du développement et pourrait jouer un rôle dans le développement des fonctions GABAergiques. Le GAT-4 n’est pas retrouvé dans les organes périphériques, ce qui fait de lui un bon candidat comme transporteur neuronal.

16
Dégradation Une fois recapté par les neurones, le GABA est recyclé ou dégradé en succinate par l’intervention successive de deux enzymes : 1. la GABA transaminase mitochondriale (3 - GABA-T), dont le cofacteur est le PLP, qui transforme le GABA en acide succinique semi-aldéhyde 2. puis, la semi-succinique aldéhyde déshydrogènase (SSA-D), dont le cofacteur est le NAD-H+, qui transforme l’acide succinique semi-aldéhyde en succinate, voie d’entrée dans le cycle de Krebs. Ces étapes de dégradation du GABA sont étroitement associées au compartiment glial.
, dont le cofacteur est le PLP, qui transforme le GABA en acide succinique semi-aldéhyde. 2. puis, la semi-succinique aldéhyde déshydrogènase (SSA-D), dont le cofacteur est le NAD-H+, qui transforme l’acide succinique semi-aldéhyde en succinate, voie d’entrée dans le cycle de Krebs. Ces étapes de dégradation du GABA sont étroitement associées au compartiment glial.")
17
Le GABA GABA GLUTAMIC ACID DECARBOXYLASE GABA TRANSAMINASE

18
LES RECEPTEURS GABAERGIQUES
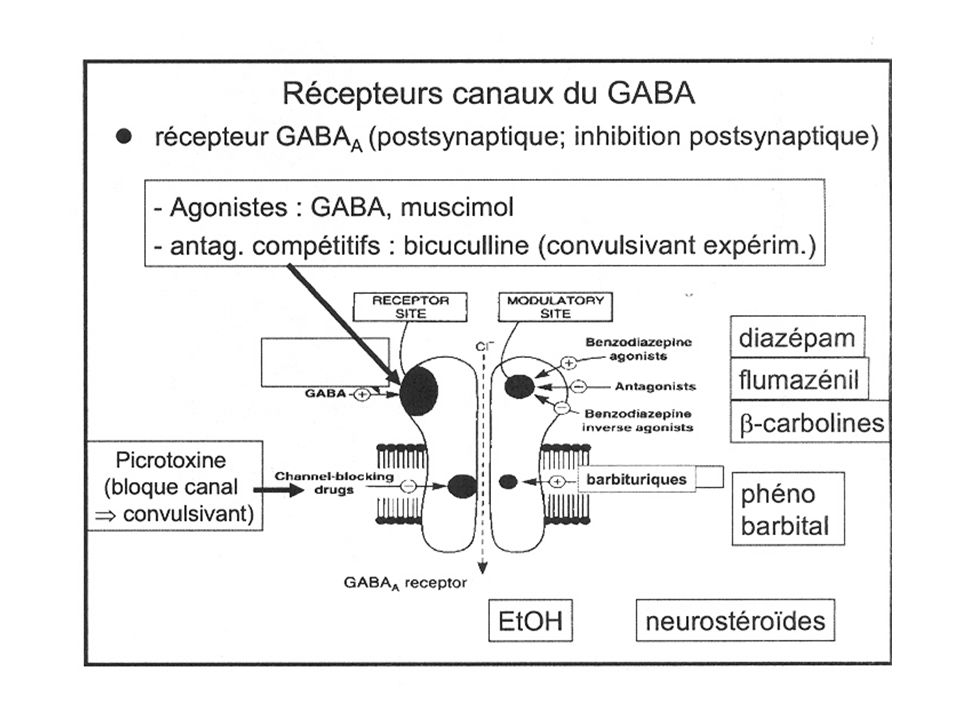
Il existe deux grands types de récepteurs GABAergiques : le récepteur GABAA et le récepteur GABAB. Le récepteur GABAA est un membre de la famille des récepteurs-canaux ioniques. Il est sensible au muscimol (agoniste) comme à la bicuculline et à la picrotoxine (antagonistes). La fixation du GABA sur son site de reconnaissance provoque l’ouverture d’un canal chlore (Cl-), qui, laissant passer les ions Cl-, produit l’hyperpolarisation de la cellule cible.
 comme à la bicuculline et à la picrotoxine (antagonistes). La fixation du GABA sur son site de reconnaissance provoque l’ouverture d’un canal chlore (Cl-), qui, laissant passer les ions Cl-, produit l’hyperpolarisation de la cellule cible.")
19
Récepteur canal Fixation de 2 molécules de GABA
Ouverture du canal chlore Entrée massive de chlore dans la cellule Hyperpolarisation de la cellule Effet global inhibiteur

20
Les récepteurs GABAA et GABAB se répartissent différemment dans le cerveau :
Les sites GABAA sont retrouvés en fortes concentrations dans le cortex cérébral, les noyaux thalamiques et la couche granulaire du cervelet. Ils sont majoritairement postsynaptiques : leur activation est responsable de potentiels postsynaptiques inhibiteurs classiques. Les sites GABAB sont retrouvés en fortes concentrations dans les couches I-III du cortex cérébral, le thalamus, les colliculus supérieurs, la couche moléculaire du cervelet et la corne dorsale de la moelle épinière. Lorsqu’ils sont situés présynaptiquement sur des terminaisons nerveuses, le GABA agit sur ces récepteurs pour réduire la libération des neurotransmetteurs contenus dans les terminaisons (diminution de la libération de noradrénaline, de glutamate, de dopamine ou de sérotonine …). Ainsi, la liaison du GABA sur ces récepteurs GABAA ou GABAB entraîne une inhibition de la neurotransmission.
. Ainsi, la liaison du GABA sur ces récepteurs GABAA ou GABAB entraîne une inhibition de la neurotransmission.")
22
Le récepteur GABAA est une glycoprotéine transmembranaire formée de 5 sous-unités, 2 alpha, 2 bêta, 1 gamma, actuellement reconnues. Il existe plusieurs types de récepteurs GABAA, différents entre eux par certaines de leurs sous-unités. On distingue actuellement 6 sous-types de sous-unités alpha, 3 sous-types de sous-unités bêta, 3 sous-types de sous-unités gamma et 1 sous-type de sous-unités delta. Ceci entraîne non seulement une grande hétérogénéité de structure mais aussi une hétérogénéité pharmacologique, dont les conséquences sont encore mal connues.

23
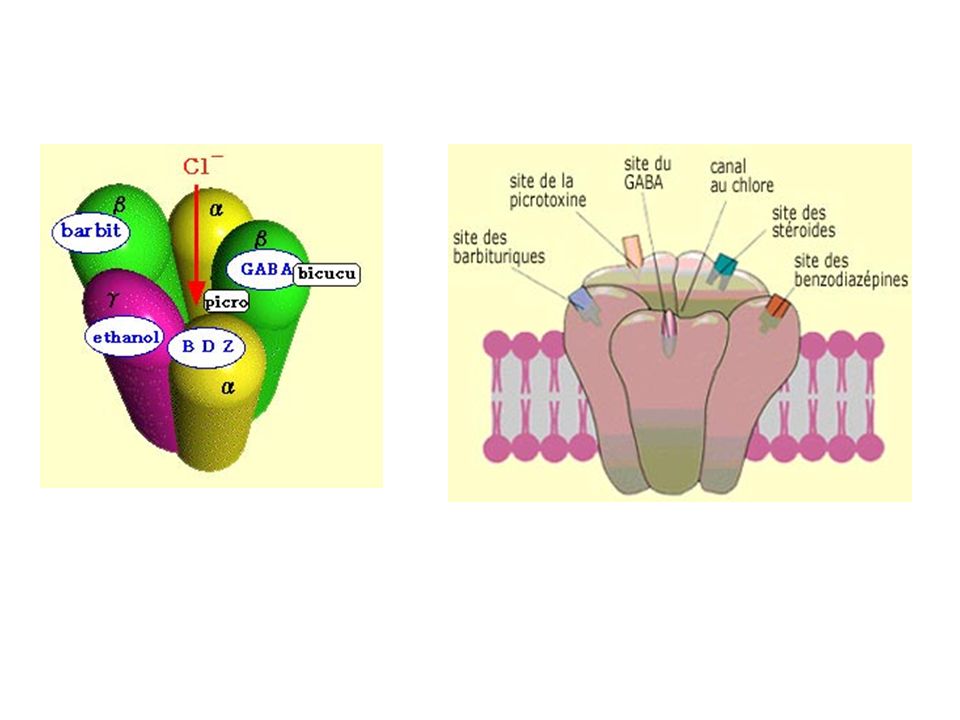
Structure moléculaire du récepteur GABA-A
5 sous-unités: 2α,2β,1γ on distingue: -6 sous-types α -3 sous-types β -3 sous-types γ Hétérogénéité de structure et hétérogénéité pharmacologique

24
Le récepteur GABAA présente, en dehors des sites récepteurs au GABA, une variété d’autres sites récepteurs topographiquement distincts capables de reconnaître des substances pharmacologiquement actives, comme les benzodiazépines (BZDs) - les barbituriques - les neurostéroïdes - les convulsivants - l'alcool ... Ces substances interagissent de manière allostérique avec les sites récepteurs au GABA et modulent la réponse GABAA.

26
Le récepteur GABAB, dont l’agoniste spécifique est le baclofène et l’antagoniste le CGP 56119, est associé à une protéine Go ou Gi (protéines liant le GTP). La protéine Go peut être couplée à un canal calcique (Ca2+ ) ou à un canal potassique (K+). Couplée à un canal Ca2+, elle entraîne une diminution des courants calciques et donc, une diminution de la libération du neurotransmetteur au niveau des terminaisons. Couplée à un canal K+, elle augmente la conductance aux ions K+ et donc, une hyperpolarisation des neurones post-synaptiques. La protéine Gi inhibe l’adénylate cyclase. Ce faisant, elle provoque une réduction intracellulaire d’AMP cyclique, ce qui peut conduire à une inhibition de la libération de neurotransmetteurs.
 ou à un canal potassique (K+). Couplée à un canal Ca2+, elle entraîne une diminution des courants calciques et donc, une diminution de la libération du neurotransmetteur au niveau des terminaisons. Couplée à un canal K+, elle augmente la conductance aux ions K+ et donc, une hyperpolarisation des neurones post-synaptiques. La protéine Gi inhibe l’adénylate cyclase. Ce faisant, elle provoque une réduction intracellulaire d’AMP cyclique, ce qui peut conduire à une inhibition de la libération de neurotransmetteurs.")
27
La plupart des benzodiazépines (BZD) sont des agonistes qui favorisent l'ouverture du canal Cl- par le GABA et ont donc un effet inhibiteur. Elles ont des propriétés pharmacologiques communes, elles ont potentiellement les mêmes indications et les mêmes effets indésirables. Il existe cependant entre les diverses BZD des différences : 1. pharmacodynamiques : certaines molécules ont un effet dominant, par exemple un effet anxiolytique ou hypnotique ou anticonvulsivant relativement plus important que les autres effets, sans que l'on en connaisse précisément l'explication. 2.pharmacocinétiques : la rapidité et la durée d'action expliquent beaucoup des différences entre molécules et leurs indications préférentielles.

28
L'adjonction de BZD au GABA potentialise l'effet du GABA - en augmentant la fréquence d'ouverture du canal Cl-. Le site récepteur au GABA serait situé dans le large domaine extra-cellulaire de la sous-unité de type bêta. Les divers hypnotiques (BZDs et apparentés) se lient donc au site récepteur des BZDs, nommé ω, faisant partie du complexe macromoléculaire GABAA - canal chlore (CMGC). Il existe toute une variété de complexes CMGC et l’on a proposé une nomenclature des différents types de récepteurs des BZDs.
 se lient donc au site récepteur des BZDs, nommé ω, faisant partie du complexe macromoléculaire GABAA - canal chlore (CMGC). Il existe toute une variété de complexes CMGC et l’on a proposé une nomenclature des différents types de récepteurs des BZDs.")
29
En effet, les études de déplacement de la liaison des BZDs dans les cellules transfectées exprimant les récepteurs GABAA contenant les différentes sous-unités alpha, associées aux sous-unités bêta et gamma, confirment l’hétérogénéité des récepteurs ω. On distingue aujourd’hui : Le récepteur BDZ1 ou ω1 Le récepteur BDZ2 ou ω2 Le récepteur BDZ3 ou ω3, essentiellement périphérique. Le cortex cérébral contient à la fois des récepteurs ω1 et ω2. Les sites ω1 sont surtout présents au niveau du cortex sensoriel et moteur. Plus fréquents dans la moelle épinière, les sites ω2 sont surtout présents au niveau des systèmes limbique (hippocampe) et extra-pyramidal.
 et extra-pyramidal.")
30
Les différences de sélectivité des BDZ et molécules apparentées à l’égard des récepteurs ω1 et ω2 peuvent expliquer les seuils d’activité très différents de leur effet hypnotique et sédatif, anxiolytique, myorelaxant et anticonvulsivant. Les BDZ «classiques», qui agissent sur les récepteurs ω1 et ω2 de façon non sélective, exercent des activités myorelaxantes et anticonvulsivantes préférentielles par rapport à leurs effets sédatifs, qui n’apparaissent qu’à doses très élevées. A l’inverse, le Zolpidem (STILNOX®), qui montre une affinité sélective pour les récepteurs ω1, a une activité sédative et hypnotique prédominante par rapport aux effets myorelaxants et anticonvulsivants, pratiquement absents.
, qui montre une affinité sélective pour les récepteurs ω1, a une activité sédative et hypnotique prédominante par rapport aux effets myorelaxants et anticonvulsivants, pratiquement absents.")
31
Les récepteurs aux barbituriques ainsi qu'à certaines hormones stéroïdes comme les dérivés de la progestérone potentialisent également la réponse du récepteur GABAA. L'effet sédatif de la progestérone, après transformation en alloprogestérone, s'expliquerait ainsi. Des stéroïdes de synthèse dits neuroactifs pourraient avoir un intérêt dans le traitement de l'épilepsie, l'insomnie et l'anxiété.

32
BZD et apparentées (Zolpidem)
Potentialisation des effets du GABA:

33
BZD et apparentées (Zolpidem)
Fixation sur les récepteurs ω Changement de conformation du récepteur GABA augmentation de l ’affinité pour le GABA Favorise l ’ouverture du canal chlore par le GABA Potentialisation des effets inhibiteurs

34
Modulation du récepteur GABAA par ses différents ligands
On distingue les agonistes, les agonistes inverses, et les antagonistes. Les agonistes augmentent la réponse au GABA. Leur activité intrinsèque est positive. Ils modulent le récepteur en augmentant la capacité du GABA à agir sur le canal chlore. Les agonistes inverses produisent l'effet opposé. Les agonistes (inverses ou non) induisent un effet pharmacologique maximal souvent avant que tous les récepteurs ne soient activés. Tous les sites de liaison du complexe macromoléculaire sont reliés allostériquement, si bien que la liaison sur une sous-unité modifie la cinétique de liaison sur les autres sous-unités.
 induisent un effet pharmacologique maximal souvent avant que tous les récepteurs ne soient activés. Tous les sites de liaison du complexe macromoléculaire sont reliés allostériquement, si bien que la liaison sur une sous-unité modifie la cinétique de liaison sur les autres sous-unités.")
35
Interaction ligand-récepteur au niveau de la synapse GABAergique

36
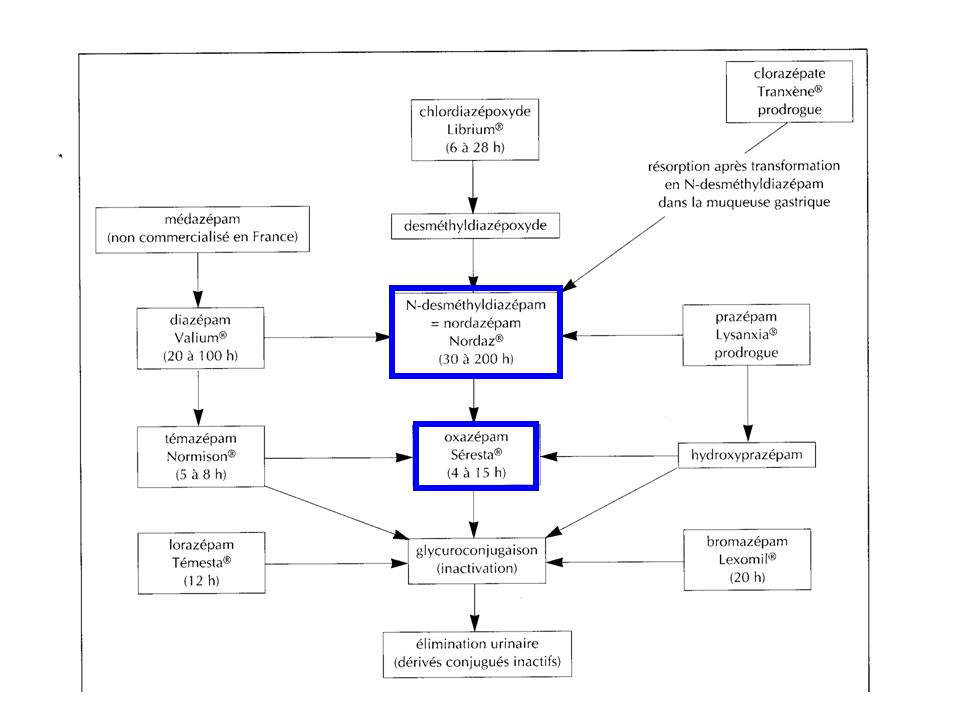
Pharmacocinétique Résorption Partie haute du tube digestif, elle est rapide et presque complète. La vitesse de résorption des BZD est le facteur limitant du délai d'action du médicament car le passage du sang au tissu cérébral est rapide. Les aliments ou les substances anticholinergiques peuvent ralentir la résorption des BZD. De même, les anti-acides ralentissent la transformation métabolique du clorazépate en desdméthyldiazépam et par conséquent sa résorption. La résorption du diazépam après administration rectale est satisfaisante.

37
Distribution Elle dépend de la liposolubilité des BZD. La liaison aux protéines plasmatiques (principalement à l'albumine) varie de 75 à 95% selon les BZD. Certaines situations sont accompagnées d'une augmentation possible des concentrations libres de BZD dans le sang (l'hypoalbuminémie, administration de contraceptifs oraux, infarctus du myocarde, repas, sevrage alcoolique).
 varie de 75 à 95% selon les BZD. Certaines situations sont accompagnées d une augmentation possible des concentrations libres de BZD dans le sang (l hypoalbuminémie, administration de contraceptifs oraux, infarctus du myocarde, repas, sevrage alcoolique).")
38
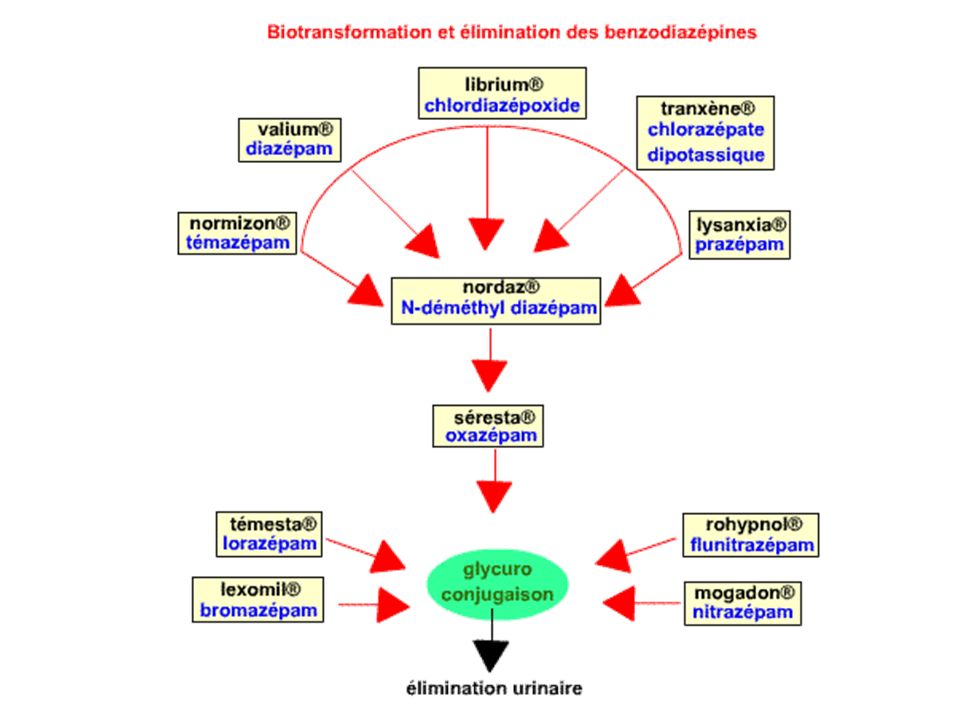
Demi-vie plasmatique : elle est beaucoup plus longue que pour les BZD hypnotiques (de 5 à 100 heures selon les molécules). Métabolisme: essentiellement hépatique, par des réactions d'oxydation microsomales (déméthylation et hydroxylation) qui donnent naissance à des composés actifs. Puis une glucuroconjugaison inactive les métabolites. Le Ndesméthyldiazépam est le principal métabolite de la plupart des produits commercialisés. Sa demi-vie est particulièrement longue et il n'atteint sa concentration d'équilibre qu'en 2 à 3 semaines. Élimination : urinaire. Les conditions physiologiques où des modifications pharmacocinétiques rencontrées sont les mêmes que pour les BZD hypnotiques (cf benzodiazépines hypnotiques).
 qui donnent naissance à des composés actifs. Puis une glucuroconjugaison inactive les métabolites. Le Ndesméthyldiazépam est le principal métabolite de la plupart des produits commercialisés. Sa demi-vie est particulièrement longue et il n atteint sa concentration d équilibre qu en 2 à 3 semaines. Élimination : urinaire. Les conditions physiologiques où des modifications pharmacocinétiques rencontrées sont les mêmes que pour les BZD hypnotiques (cf benzodiazépines hypnotiques).")
41
Volume de distribution(I/kg)
DCI Biodisponibilité Demi-vie(h) Liaison protéine(%) Volume de distribution(I/kg) Alprazolam 80-90 10-15 70-80 1 Bromazépam 84 20 70 Chlordiazépoxyde - 5-30 94-97 0,3-0,6 Clobazam 9-30 88-92 Clorazépate 33-55 97 1,60 Clotiazépam 3,5-5 98 Desméthyldiazépam Nordazépam Nordiazépam 50-120 94-98 0,9-1,3 Diazépam 20-70 97-99 0,9-2 Loflazépate 50-100 0,7 Lorazépam 91-95 10-20 07-1 Oxazépam 5-15 95-97 0,6 Prazépam 80-100 1,40 Tofisopam 8
 Liaison protéine(%) Volume de distribution(I/kg) Alprazolam Bromazépam Chlordiazépoxyde ,3-0,6. Clobazam Clorazépate ,60. Clotiazépam. 3, Desméthyldiazépam. Nordazépam. Nordiazépam ,9-1,3. Diazépam ,9-2. Loflazépate ,7. Lorazépam Oxazépam ,6. Prazépam ,40. Tofisopam. 8.")
42
insomnies d'endormissement;
Indications anxiété réactionnelle, notamment les troubles de l'adaptation avec humeur anxieuse et l'anxiété post-traumatique; anxiétés généralisées; insomnies d'endormissement; traitement d'appoint de l'anxiété au cours des névroses (hystérie, hypocondrie, phobie); crise d'angoisse; certaines formes de tremblements; prévention et traitement du delirium tremens; désintoxication alcoolique (en cure courte); attaque de panique (alprazolam); prévention et traitement des crises convulsives du nourrisson et de l'enfant (diazépam).
; crise d angoisse; certaines formes de tremblements; prévention et traitement du delirium tremens; désintoxication alcoolique (en cure courte); attaque de panique (alprazolam); prévention et traitement des crises convulsives du nourrisson et de l enfant (diazépam).")
43
DCI Spécialités Présentation (mg) Posologie moyenne adulte (mg/j) Nombre de prise par jour alprazolam Xanax® cp. sec 0,25 0,5 à 4 2 à 3 bromazépam lexomil® cp. 6 quadrisécable 3 à 12 1 à 3 chlordiazépoxyde librium® cp. 5 et gel. 10 15 à 40 clo bazam Urbanyl® gel. 5, cp. sec. 10 et 20 10 à 60 clorazépate Tranxène® gel. 5 et 10, cp. sec 50 5 à 100 1 à 2 clotiazépam Vératran® cp. sec. 5 et 10 10 à 30 diazépam Valium® cp. 2, 5 et 10 5 à 20 Novazam® cp. 10 quadrisécable esta zolam Nuctalon ® cp. sec 2 au coucher flunitrazépam Rohypnol® cp. sec 1 et 2 0,5 à 2 Noriel® loflazépate Victan® cp. sec. 2 2 loprazolam Havlane® cp. sec 1 0,5 à 1 lorazépam Témesta® cp. 1 et 2,5 2 à 5 lormét azépam Noctamid® 0,5 à2 nitrazépam Mogadon® cp. sec 5 2,5 à 10 nordazépam Nordaz® cp. sec 7,5 et 7,5 à 15 1 au coucher cp. 15 quadrisécable oxazépam Séresta® cp. 10 et 50 30 à 60 3 à 4 prazépam lysanxia® cp. 10 et gtte 20 à 40 1 à 2 témazépam Normison® caps. 10 et 20 mg 10 à 20 au coucher tofisopam Sériel® cp.50 100 à 300 3 à 4 triazolam Halcion® cp. séc. 0,125 mg 0,125 au coucher

44
Effets indésirables Variables en fonction de la posologie, la sensibilité individuelle et la T1/2: somnolence diurne, difficulté de concentration, sensation ébrieuse; phénomène de tolérance : après 2 à 3 mois d'utilisation; syndrome de sevrage et effet rebond à l'arrêt brutal du traitement à doses élevées : somnolence et quelquefois coma à doses toxiques avec dépression respiratoire; troubles de la mémoire (amnésie antérogrades) et troubles confusionnels (attention chez les sujets âgés)
 et troubles confusionnels (attention chez les sujets âgés)")
45
Surdosage Le plus à craindre est la dépression respiratoire. Il existe un antidote aux BZD, le flumazénil (Anexate®). Antagoniste des benzodiazépines, il bloque spécifiquement par inhibition compétitive les effets sur le SNC exercés par les substances agissant au niveau des récepteurs aux BZD. La t1/2 courte du flumazénil, inférieure à celle de la plupart des BZD impose un traitement d’entretien et ne dispense pas d’une surveillance intensive en raison des risques de réendormissement secondaires et de convulsions notamment en cas de prise associée avec d’autres toxiques ( alcool, ADTC). Si l’intoxication fait suite à un traitement chronique aux BZD, le flumazénil peut conduire à un syndrome de sevrage. On administre alors de faibles doses de BZD.
. Antagoniste des benzodiazépines, il bloque spécifiquement par inhibition compétitive les effets sur le SNC exercés par les substances agissant au niveau des récepteurs aux BZD. La t1/2 courte du flumazénil, inférieure à celle de la plupart des BZD impose un traitement d’entretien et ne dispense pas d’une surveillance intensive en raison des risques de réendormissement secondaires et de convulsions notamment en cas de prise associée avec d’autres toxiques ( alcool, ADTC). Si l’intoxication fait suite à un traitement chronique aux BZD, le flumazénil peut conduire à un syndrome de sevrage. On administre alors de faibles doses de BZD.")
46
Précautions d'emploi - prudence chez les conducteurs de machines, chez les sujets âgés, les éthyliques; - chez l'enfant la prescription doit être ponctuelle et de courte durée - ne pas associer à l'alcool; - éviter en cas de grossesse ou d'allaitement; - en cas de traitement prolongé ou d'utilisation à doses élevées, prescrire de faibles doses pendant 10 semaines au maximum et arrêter de façon progressive sur 2 semaines; - insuffisance rénale et/ou hépatique: il peut être nécessaire d'adapter la posologie; - en cas d'insuffisance respiratoire modérée ou de myasthénie, la posologie doit être diminuée et l'administration nécessite une surveillance étroite.

47
Sevrage aux benzodiazépines
L'arrêt du traitement peut entraîner un phénomène de sevrage. Le patient en sera averti. L’arrêt sera donc progressif avec décroissance posologique sur plusieurs semaines, d'autant plus que l'utilisation a été prolongée, où que l'on suspecte une pharmacodépendance. Contre-indications Insuffisances respiratoires sévères, allergie aux BZD. Limitation de la durée de prescription L'arrêté du 7 octobre 1991 a réduit la durée de prescription des médicaments contenant des substances de la liste I à propriétés hypnotiques et/ou anxiolytiques respectivement à 4 semaines pour les hypnotiques et 12 semaines pour les anxiolytiques. Le triazolam (Halcion®) a une durée de prescription limitée à 2 semaines.
 a une durée de prescription limitée à 2 semaines.")
48
MEPROBAMATE (famille des carbamates)
Les carbamates utilisées en thérapeutique représentent l’ancienne génération. Ce sont des esters d’alcool et d’acide carbamique. Demeure le méprobamate dont le mécanisme d’action est probablement proche de celui des BDZ. Pharmacocinétique absorption: au niveau du tractus gastrointestinal; distribution: liaison aux protéines plasmatiques d'environ 20%. On observe un passage dans le lait et à travers la barrière placentaire; demi-vie plasmatique: 6 à 16 heures; métabolisme: hépatique (hydroxylation, glucuroconjugaison); élimination : urinaire;
; élimination : urinaire;")
49
Indications: molécules de moins en moins utilisées
- anxiété excessive, insomnie d'endormissement; - syndrome de sevrage éthylique (usage français); Les formes injectables sont utilisées dans les états d'agitation aigus, délirium tremens, crises d'angoisse aiguës, prémédication avant certains examens. Effets indésirables: somnolence diurne, sensation ébrieuse, irritabilité; tolérance et dépendance avec syndrome de sevrage à l'arrêt du traitement lors d'utilisation à fortes doses. En cas de surdosage cad pour des concentrations plasmatiques supérieures à 50 mg/l, traiter par une diurèse osmotique ou une épuration extrarénale et en cas de défaillance cardiaque (concentrations supérieures à 150 mg/l), utiliser la dobutamine (Dobutrex®). Précautions d'emploi: sujets âgés ou myasthéniques. Contre-indications : insuffisances respiratoires sévères, grossesse (premier trimestre) et allaitement, porphyries aiguës intermittentes.
; Les formes injectables sont utilisées dans les états d agitation aigus, délirium tremens, crises d angoisse aiguës, prémédication avant certains examens. Effets indésirables: somnolence diurne, sensation ébrieuse, irritabilité; tolérance et dépendance avec syndrome de sevrage à l arrêt du traitement lors d utilisation à fortes doses. En cas de surdosage cad pour des concentrations plasmatiques supérieures à 50 mg/l, traiter par une diurèse osmotique ou une épuration extrarénale et en cas de défaillance cardiaque (concentrations supérieures à 150 mg/l), utiliser la dobutamine (Dobutrex®). Précautions d emploi: sujets âgés ou myasthéniques. Contre-indications : insuffisances respiratoires sévères, grossesse (premier trimestre) et allaitement, porphyries aiguës intermittentes.")
50
Produits utilisés (liste I): formes et posologies
: formes et posologies")
51
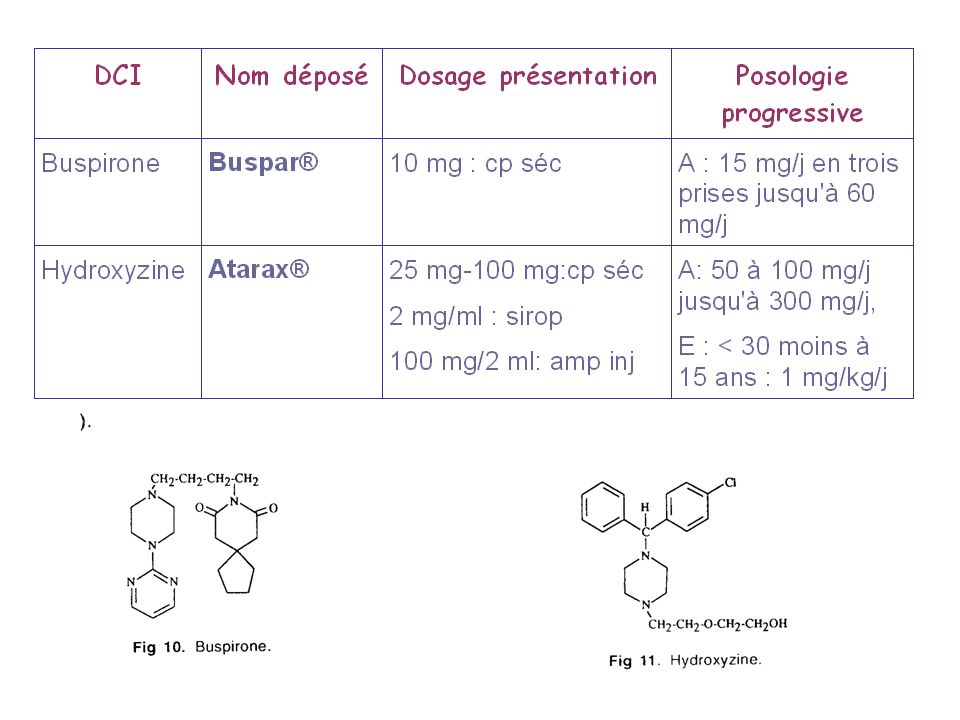
ANXIOLYTIQUES DIVERS Buspirone : Buspar ®(liste I).
Il s'agit du seul représentant de la famille des pipéridyl-pipérazines, c'est un anxiolytique pur dépourvu des effets indésirables des BZD. Il n'altère pas les capacités physiques et psychiques des patients. La buspirone est un antagoniste sérotoninergique. Pharmacocinétique - Résorption : rapide, pic plasmatique atteint en 60 à 90 minutes. La concentration à l'état d'équilibre est atteinte au bout de 2 jours; - Distribution : la fixation aux protéines est d'environ 95%; - Demi-vie d’élimination : 2 à 11 heures; - Métabolisme : hépatique; - Élimination : urinaire (60%) et biliaire (40%). Contre-indiqué en cas d'insuffisances rénale ou hépatique sévères.
 et biliaire (40%). Contre-indiqué en cas d insuffisances rénale ou hépatique sévères.")
52
Hydroxyzine : Atarax (liste I).
Dérivé de la pipérazine, c'est un anxiolytique et sédatif ayant un effet anti-allergique. Pharmacocinétique - Résorption : rapide par le tractus gastro-intestinal; - Distribution : le taux plasmatique maximal est obtenu en 2 h à 2 h 30; le délai d'action après prise per os est de 15 à 30 minutes, la durée d'action est de l'ordre de 6 à 8 heures. - Indications : il est prescrit contre les insomnies d'endormissement, les réactions allergiques (prurit, éruption, urticaire) et en prémédication avant les interventions. - Effets indésirables : bouche sèche, constipation, troubles de l’accommodation. A éviter en cas de grossesse (premier trimestre), de glaucome, d'adénome prostatique.
 et en prémédication avant les interventions. - Effets indésirables : bouche sèche, constipation, troubles de l’accommodation. A éviter en cas de grossesse (premier trimestre), de glaucome, d adénome prostatique.")
55
Cl Cl Cl

Présentations similaires