Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Immunopathologie Michel Moutschen

2
Plan du cours 1. Immunodéficiences primaires
2. VIH et immunodéficiences secondaires 3. Atopie 4 .Autoimmunité (principes généraux) 5. Autoimmunité (grands syndromes cliniques) 6. Inflammation 7. Transplantation et immunosuppresseurs
 5. Autoimmunité (grands syndromes cliniques) 6. Inflammation. 7. Transplantation et immunosuppresseurs.")
3
Immunodéficience

4
Signes d’appel évoquant une immunodéficience

5
Signes d’appel Infections fréquentes Infections prolongées
Infections dans des sites inhabituels Infections à germes inhabituels Septicémies, méningites à répétition Histoire familiale

7
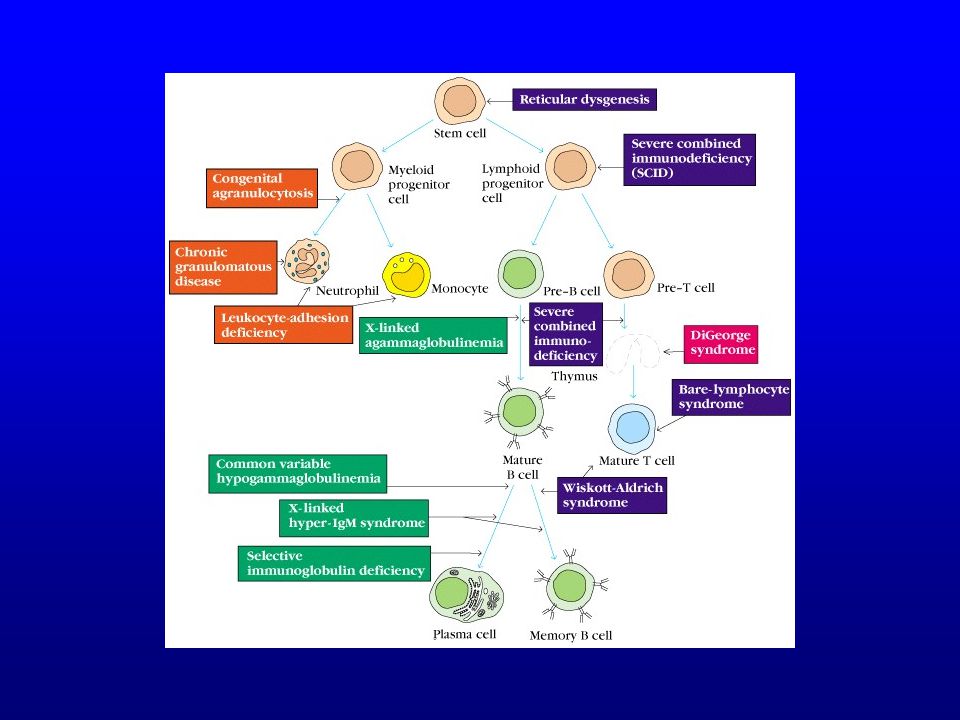
Groupes étiologiques Primitifs (génétiques)
typiquement affection pédiatrique avec histoire familiale mais : déficits génétiques parfois sporadiques (néomutations) apparition éventuelle à l’âge adulte (expression variable) hérédité parfois polygénique faisant intervenir facteurs de l’environnement
 apparition éventuelle à l’âge adulte (expression variable) hérédité parfois polygénique faisant intervenir facteurs de l’environnement.")
8
Groupes étiologiques (2)
Acquis : intervention prépondérante d’un phénomène extrinsèque agent infectieux problème métabolique (diabète, malnutrition) médicament (dans le cadre d’une transplantation ou d’une maladie autoimmunitaire) cancer splénectomie
 médicament (dans le cadre d’une transplantation ou d’une maladie autoimmunitaire) cancer. splénectomie.")
9
Compartiment touché et type d’infection
Immunité humorale germes pyogènes encapsulés (pneumocoques, Hemophilus influenzae, staphylocoques) virus généralement primoinfection normale mais déficit de l’immunité à long terme (plusieurs poussées de zona, de rougeole,...) atteintes neurologiques à entérovirus (coxsackies, poliovirus) parasites : giardiases intestinales
 virus. généralement primoinfection normale mais déficit de l’immunité à long terme (plusieurs poussées de zona, de rougeole,...) atteintes neurologiques à entérovirus (coxsackies, poliovirus) parasites : giardiases intestinales.")
10
Types d’infection Immunité à médiation cellulaire
infections mycotiques (candidose mucocutanée,…) infections virales (svt virus latents de la famille herpès) pneumocystoses bactéries intracellulaires (mycobactéries) svt déficits qualitatifs de l’immunité humorale
 infections virales (svt virus latents de la famille herpès) pneumocystoses. bactéries intracellulaires (mycobactéries) svt déficits qualitatifs de l’immunité humorale.")
11
Types d’infection Complément Phagocytes
septicémies et méningites à germes encapsulés Phagocytes infections de la peau et du système réticulo-endothélial (ganglions, rate) Déficits immunitaires combinés (sévères) : SCID et CID infections très sévères, d’abord de type «T », puis de type « B »
 Déficits immunitaires combinés (sévères) : SCID et CID. infections très sévères, d’abord de type «T », puis de type « B »")
12
Immunodéficiences primitives
plus de 70 formes décrites déficits souvent très rares déficit de l ’immunité humorale dans 70% des cas les formes les plus fréquentes : déficit en IgA (1:400 à 1:800) hypogammablobulinémie commune variable déficit sélectif en sous-classes
 hypogammablobulinémie commune variable. déficit sélectif en sous-classes.")
13
Immunodéficiences primitives
progrès considérable dans leur compréhension intérêt physiopathologique classification OMS : (l’adresse du site est toujours correcte, consultation utile, bonne synthèse)
")
15
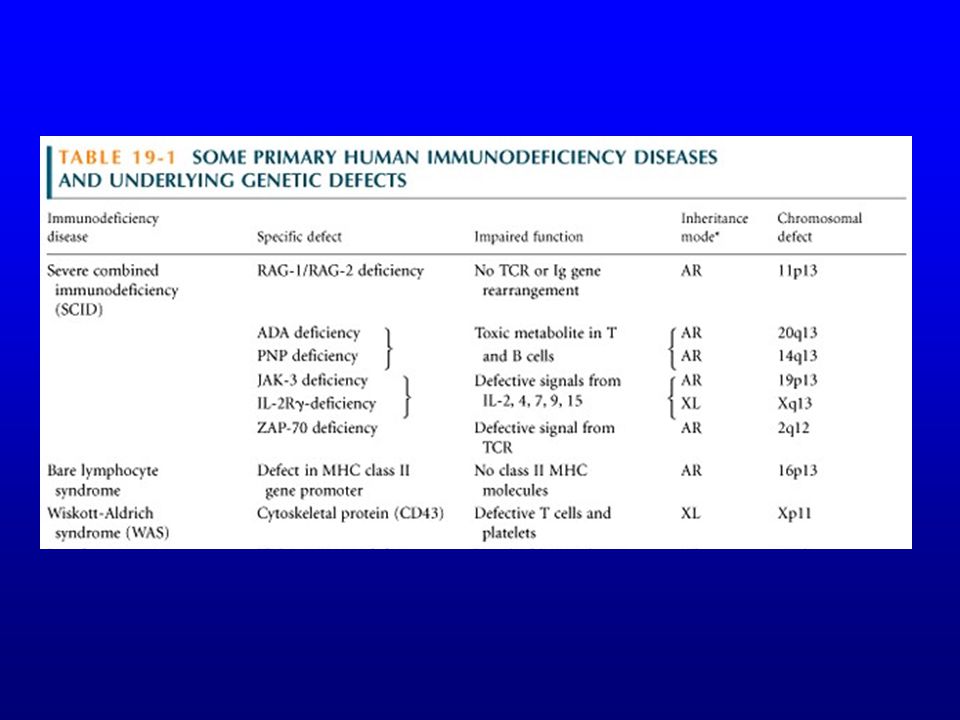
Classification SCID (severe combined immunodeficiency)
Hypogammaglobulinémies Déficits « purs » des lymphocytes T Déficit des cellules phagocytiques Déficit de facteurs du complément

16
SCID : déficits immunitaires combinés (B+T) sévères
 sévères")
17
SCID Plusieurs types de SCID
Toujours déficit majeur de l’immunité cellulaire Déficit secondaire de l’immunopoïèse B (maturation d’affinité, commutation isotypique) souvent mais pas toujours d’ agammaglobulinémie associée
 souvent mais pas toujours d’ agammaglobulinémie associée.")
18
SCID Présentation Infections TRES précoces (après la naissance)
Virales (RSV, CMV, entérovirus, parainfluenza) Parasitaires (Pneumocystis carinii) Mycotiques (Candida) Bactériennes : pneumonies, otites, septicémies (plus tard)
 Parasitaires (Pneumocystis carinii) Mycotiques (Candida) Bactériennes : pneumonies, otites, septicémies (plus tard)")
20
SCID AR T-B- (20%) AR T-B+ X-linked T-B+ (60%)
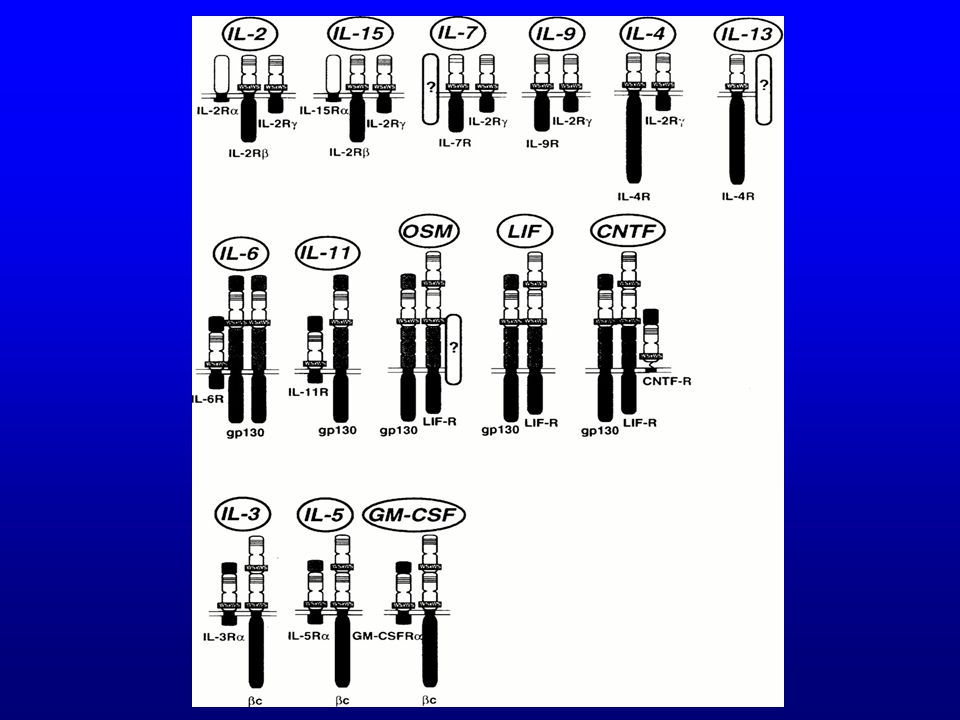
Gènes de la chaîne légère de substitution (V pré-B et l5); RAG-1 et RAG-2 AR T-B+ Svt déficit JAK-3 X-linked T-B+ (60%) Déficit de la chaîne g commune aux récepteurs de l ’IL-2, IL-4, IL-7, IL-9 et IL-15
; RAG-1 et RAG-2. AR T-B+ Svt déficit JAK-3. X-linked T-B+ (60%) Déficit de la chaîne g commune aux récepteurs de l ’IL-2, IL-4, IL-7, IL-9 et IL-15.")
21
Déficit de la chaîne g du récepteur de l’interleukine-2

23
Autres causes de SCID Déficit en ADA (Adénosine désaminase) (20%)
Effets toxiques du dATP et de la S-adénosyl homocystéine Déficit en produits MHC de classe I ou de classe I & II (syndrome des lymphocytes nus) Afrique du Nord, hypogammaglobulinémie + lymphopénie CD4 (si déficit classes II)
 Afrique du Nord, hypogammaglobulinémie + lymphopénie CD4 (si déficit classes II)")
24
Hypogammaglobulinémies (I)
Hypogammaglobulinémie liée au X (XLA-maladie de Bruton) Hypogammaglobulinémie commune variable (CVI) Déficit sélectif en IgA Déficit sélectif en sous-classes Déficits qualitatifs
 Hypogammaglobulinémie commune variable (CVI) Déficit sélectif en IgA. Déficit sélectif en sous-classes. Déficits qualitatifs.")
25
Hypogammaglobulinémies (II)
Hypergammaglobulinémie M liée au X Déficit sélectif en IgM Syndrome lymphoprolitératif lié au (syndrome de Duncan) Syndrome d’hypergammaglobulinémie E (syndrome de Job) Hypogammaglobulinémie transitoire du jeune enfant
 Syndrome d’hypergammaglobulinémie E (syndrome de Job) Hypogammaglobulinémie transitoire du jeune enfant.")
26
Hypogammaglobulinémies
2’ : agammaglobulinémie autosomale récessive 4’ : SCID (rag-1 et rag-2) 7’ : (chaîne légère de substitution du préBCR) 9’ : XLA (tyrosine kinase de Bruton)
 7’ : (chaîne légère de substitution du préBCR) 9’ : XLA (tyrosine kinase de Bruton)")
27
tyrosine kinase de Bruton

28
XLA (Bruton) Etiologie tyrosine kinase de Bruton (PTK famille TEK)
Xq délétions et mutations ponctuelles pénétrance variable mutations fréquentes

29
XLA (II) Présentation garçons de plus de 6 mois (avec ou sans histoire familiale) infections pulmonaires et ORL à répétition Hemophilus influenzae Streptococcus pneumoniae méningites arthrites septiques Staphylococcus aureus

30
XLA (III) Complications tardives Bronchectasies
Méningoencéphalite chronique (echovirus et coxsackies) Arthrites à Ureaplasma urealyticum
 Arthrites à Ureaplasma urealyticum.")
31
XLA (IV) Diagnostic IgA, G, M, D et E absentes ou effondrées
lymphocytes B absents ou très rares (CD20) pas de centres germinatifs dans les ganglions, pas d’amygdales lymphocytes T qualitativement ou quantitativement normaux déficit expression de BTK (cytofluorimétrie ou westernblot)
 pas de centres germinatifs dans les ganglions, pas d’amygdales. lymphocytes T qualitativement ou quantitativement normaux. déficit expression de BTK (cytofluorimétrie ou westernblot)")
32
Hypogammaglobulinémie commune variable (CVID)
Etiologie ? Histoire familiale de CVI, de déficits en IgA, de déficits en sous-classe histoire familiale de maladies autoimmunes A1B8C4DR3DQ0 Probablement déséquilibre de linkage avec allèle de classe III Hétérogène

33
CVID Présentation Tout âge (svt jeunes enfants ou jeunes adultes)
Infections bactériennes récurrentes Autoimmunité (thyroïde, diabète, vitiligo, pelade) Hyperplasie nodulaire lymphoïde de l’intestin Granulomes pulmonaires ou hépatiques
 Hyperplasie nodulaire lymphoïde de l’intestin. Granulomes pulmonaires ou hépatiques.")
34
CVID (III) Complications tardives
Infections : bronchectasies, cholangites à Campylobacter, arthrites à Ureaplasma urealyticum Malabsorption (entéropathie de type maladie coeliaque) Splénomégalie, hypersplénisme Lymphomes Autoimmunité Thymome (myasthénie grave, anémie aplastique, PTI)
 Splénomégalie, hypersplénisme. Lymphomes. Autoimmunité. Thymome (myasthénie grave, anémie aplastique, PTI)")
35
CVID (IV) Diagnostic Ig abaissées de façon variable
Déficit qualitatif toujours présent Lymphocytes B normaux ou abaissés Lymphopénie T (CD4+ CD45RA+) Anergie T fréquente
 Anergie T fréquente.")
36
Déficit en IgA

37
IgA Présente dans le sérum (15% des Ig) mais surtout importante par sa présence dans les sécrétions digestives respiratoires génito-urinaires collostrum larmes

38
IgA la forme sérique est monomérique la forme sécrétée est polymérique
chaîne S pièce sécrétoire

39
Sécrétion des IgA

40
Déficit en IgA Très fréquent : 1/400 à 1/800 Etiologie
Contexte génétique Famille de CVID B8DR3 Le plus souvent déficit des IgA sériques & des IgA sécrétoires (problème au niveau de la synthèse) Très rarement : déficit de pièce sécrétoire (rarissime)
 Très rarement : déficit de pièce sécrétoire (rarissime)")
41
Déficit en IgA (II) Présentation Décours souvent bénin
Infections ORL fréquentes, rarement sévères Phénomènes allergiques et autoimmunitaires (LED, PR, JCA, Biermer) Réactions transfusionnelles
 Réactions transfusionnelles.")
42
Déficit en IgA (III) Diagnostic
IgA sériques indétectables (<0.05g/l) Pas d’IgA salivaire, présence d’IgG et d’IgM IgG totales et IgM normales, IgG2 et IgG4 svt abaissées IgE svt accrues Autoanticorps fréquents dont anti-IgA Fonction T normale
 Pas d’IgA salivaire, présence d’IgG et d’IgM. IgG totales et IgM normales, IgG2 et IgG4 svt abaissées. IgE svt accrues. Autoanticorps fréquents dont anti-IgA. Fonction T normale.")
43
Déficit sélectif en sous-classes d’IgG
Etiologie ? Même famille que CVID et déficit IgA (B8DR3) Attention : variations allotypiques (allotypes Gm) et raciales des concentrations des différentes sous-classes
 Attention : variations allotypiques (allotypes Gm) et raciales des concentrations des différentes sous-classes.")
44
Classes et sous-classes

45
Déficit sélectif en sous-classes d’IgG (II)
Présentation Tout âge IgG2 et/ou IgG4 : infections bactériennes récurrentes, bronchectasies IgG3 : sinusites, asthme (mais épiphénomène?)
")
46
Déficit sélectif en sous-classes d’IgG (III)
Diagnostic Déficit partiel ou total (p.ex. IgG4) (tenir compte de l’âge et de l’appartenance ethnique pour l’interprétation IgG normales ou discrètement abaissées (si déficit IgG1), IgA nles ou abaissées, IgM nles Lymphocytes B et T normaux Déficit qualitatif fréquent
 (tenir compte de l’âge et de l’appartenance ethnique pour l’interprétation. IgG normales ou discrètement abaissées (si déficit IgG1), IgA nles ou abaissées, IgM nles. Lymphocytes B et T normaux. Déficit qualitatif fréquent.")
47
Déficit sélectif avec concentration normale d’Ig SPAD
Etiologie ? Déficit préférentiel des anticorps dirigés contre les polysaccharides (T-indépendants) (p.ex. pneumocoque) et certaines protéines p.ex. HBV Lymphocytes B et T quantitativement normaux Fréquence accrue de lymphocytes B CD5+ ?
 (p.ex. pneumocoque) et certaines protéines p.ex. HBV. Lymphocytes B et T quantitativement normaux. Fréquence accrue de lymphocytes B CD5+")
48
Hypogammaglobulinémies rares
X-linked hyperIgM Déficit de commutation isotypique (IgM et IgD élevées, IgG, A et E basses) Déficit en CD40L sur lymphocytes T Anomalies fonctionnelles des lymphocytes T Infections bactériennes et parasitaires (Pneumocystis carinii, Cryptosporidium) Autoimmunité, lymphomes B
 Déficit en CD40L sur lymphocytes T. Anomalies fonctionnelles des lymphocytes T. Infections bactériennes et parasitaires (Pneumocystis carinii, Cryptosporidium) Autoimmunité, lymphomes B.")
49
gp39=CD40L=CD154

50
Syndrome lymphoprolifératif lié au X (Duncan-XLP)
Infection EBV gravissime Primo-infection fatale Lymphome Immunodéficience sévère Gène codant pour une protéine à domaine SH2 (DSHP « SHIP » like?)
")
51
Hypogammaglobulinémie transitoire de l’enfant
Fréquente dans les familles où existent des hypogammaglobulinémies 6 mois à 36 mois Valeurs basses d’IgG et d’IgA, IgM nles Nombre normal de lymphocytes B

52
Syndrome hyper-IgE (Job)
Eczéma atypique, infections cutanées invasives et récurrentes, ostéopénie, fractures Anomalie fonctionnelle des lymphocytes T et B. Immunodéviation clonale sur un mode Th2 (ou Th0?) IgE très accrues > kU/l Déficit secondaire des polynucléaires (effet inhibiteur des IgE)
 IgE très accrues > kU/l. Déficit secondaire des polynucléaires (effet inhibiteur des IgE)")
53
Déficit des lymphocytes T

54
Déficit des lymphocytes T
DiGeorge (Wiskott-Aldrich) (Ataxie-télangiectasie)
 (Ataxie-télangiectasie)")
55
DiGeorge Spectre de nombreuses anomalies du développement. Plus qu’un gène impliqué. Anomalies des structures dérivées de l’arc branchial (cœur, thymus, parathyroïdes, face) Notamment Catch 22 (microdélétion 22q11) Déficit d’une protéine en doigt de zinc Autres syndromes similaires Délétion 10p, Sprintzen, VCF, Kallman,… Syndrome foeto-alcoolique, diabète maternel, embryopathie aux rétinoïdes
 Notamment Catch 22 (microdélétion 22q11) Déficit d’une protéine en doigt de zinc. Autres syndromes similaires. Délétion 10p, Sprintzen, VCF, Kallman,… Syndrome foeto-alcoolique, diabète maternel, embryopathie aux rétinoïdes.")
56
a1hkaz

57
DiGeorge (II) Présentation Tétanie néonatale
Malformations cardiaques (tronc artériel, Fallot,…) Fente palatine, implantation basse des oreilles, bouche « de poisson » Réduction partielle ou totale du thymus Immunodéficience qui s’améliore avec l’âge GVHD si transfusions
 Fente palatine, implantation basse des oreilles, bouche « de poisson » Réduction partielle ou totale du thymus. Immunodéficience qui s’améliore avec l’âge. GVHD si transfusions.")
58
DiGeorge (III) Diagnostic Lymphopénie T d’importance variable
Réponses fonctionnelles des lymphocytes T normales ou diminuées Ig normales ou légèrement diminuées. Déficit qualitatif Cytogénétique

59
Déficits phagocytaires
Maladies granulomateuses chroniques (CGD) Liée au X AR Déficit d’adhésion des leucocytes (LAD) LAD-1 LAD-2 Déficit en G6PD Déficit en myélopéroxydase Chediak-Higashi
 Liée au X. AR. Déficit d’adhésion des leucocytes (LAD) LAD-1. LAD-2. Déficit en G6PD. Déficit en myélopéroxydase. Chediak-Higashi.")
60
CGD Déficit de bactéricidie intracellulaire (explosion respiratoire insuffisante) Infections à germes catalase+ (Staph. aureus, Aspergillus, Nocardia, Serratia) avec abcès profonds (notamment hépatiques) et granulomes chroniques Déficit de composants du cytochrome b558 (X) ou de la NADPH oxydase (AR) Diagnostic : réduction du nitrobleu (NBT)
 avec abcès profonds (notamment hépatiques) et granulomes chroniques. Déficit de composants du cytochrome b558 (X) ou de la NADPH oxydase (AR) Diagnostic : réduction du nitrobleu (NBT)")
61
LAD LAD-1 Déficit de la chaîne b commune à CD18, LFA-1, MAC-1 et CR4
Infections intestinales et cutanées; périodontites Pas d ’inflammation locale, pas de pus Séparation tardive du cordon ombilical Neutrophilie accrue Diagnostic : Cytofluorimétrie

62
LAD

63
Complément

64
Complément

65
Déficits du complément

66
Déficit inhibiteur de C1 estérase
AD 2 formes : I (pas de protéine) et II (perte de l’activité) Inactivation insuffisante de la cascade du complément et de celle des kinines Angioedème (œdème profond sans urticaire, sans prurit) Diagnostic : dosage de l ’inhibiteur de C1 esterase, C2, C4
 et II (perte de l’activité) Inactivation insuffisante de la cascade du complément et de celle des kinines. Angioedème (œdème profond sans urticaire, sans prurit) Diagnostic : dosage de l ’inhibiteur de C1 esterase, C2, C4.")
67
Exploration des déficits immunitaires
screening sang complet et formule hémoleucocytaire dosage immunoglobulines IgG, A, M, D, E 2ème ligne typage lymphocytaire marqueurs T (CD3, CD4, CD8 marqueurs B (CD19, CD20, CD21, Ig) marqueurs NK (CD16) marqueurs d’activation (HLA-DR, CD25)
 marqueurs NK (CD16) marqueurs d’activation (HLA-DR, CD25)")
68
Exploration des déficits immunitaires (II)
2ème ligne évaluation fonctionnelle T (prolifération in vitro, DTH) évaluation fonctionnelle B après vaccination, réponse humorale contre antigène protéique (p.ex. tétanos) ou polysaccharidique (pneumocoque) sous-classes IgG (IgG2, IgG3 et IgG4)
 évaluation fonctionnelle B. après vaccination, réponse humorale contre antigène protéique (p.ex. tétanos) ou polysaccharidique (pneumocoque) sous-classes IgG (IgG2, IgG3 et IgG4)")
69
Exploration des déficits immunitaires (III)
2ème ligne complément CH50 (classique et alterne) C3 et C4 Phagocytes phagocytose, réduction du nitrobleu, chimiotactisme
 C3 et C4. Phagocytes. phagocytose, réduction du nitrobleu, chimiotactisme.")
70
Prise en charge générale des patients présentant des déficits immunitaires
mesures générales d’hygiène éliminer allergènes si atopie kiné respiratoire éviter vaccins vivants (polio, fièvre jaune)
")
71
Prise en charge générale des patients présentant des déficits immunitaires
vaccin antigrippal et antipneumococcique antibiothérapies précoces traitements spécifiques immunoglobulines IV pour les déficits en IgG avec répercussion clinique peu de place pour les immunoglobulines IM et pour les lysats bactériens oraux (bronchovaxom)
")
72
Traitement des hypogammaglobulinémies
Immunoglobulines par voie intraveineuse Toutes les 3 à 4 semaines Viser une concentration résiduelle d’IgG supérieure à 5g/l Contre-indication dans les déficits sélectifs en IgA avec présence d’anti-IgA

73
Traitement des hypogammaglobulinémies
Dans les déficits en IgA sans anti-IgA et les déficits sélectifs en IgG2, surveiller régulièrement l’apparition des anti-IgA Pas un traitement anodin cher risque faible mais non nul de transmission d ’agents infectieux (HCV) effets secondaires occasionnels
 effets secondaires occasionnels.")
74
Immunodéficiences acquises
Infections HIV Autres infections virales: EBV, CMV, rubéole, rougeole, influenza Autres infections (bactériennes, mycotiques, parasitaires)
")
Présentations similaires

>")


















