Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
DIAGNOSTIC D’UN ALLONGEMENT DU TEMPS DE SAIGNEMENT
ygfu LUANGKHOT Elodie PUTIN Cyril DES Hématologie 21 Novembre 2006

2
PHYSIOLOGIE DE L’HEMOSTASE PRIMAIRE

3
Généralités Fonction hémostatique normale assure
Maintien du sang à l’état liquide Maintien de la masse sanguine En cas de brèche vasculaire : processus localisés, rapides et non extensifs permettent la réparation d’une lésion vasculaire et l’arrêt d’un saignement

4
Généralités Hémostase : étapes inter-dépendantes au déroulement concomitant Hémostase primaire : interaction plaquettes-vaisseaux Clou plaquettaire ou thrombus blanc Coagulation : transformation du fibrinogène en fibrine Caillot de fibrine ou thrombus rouge Fibrinolyse : phénomène lent d’expression tardive Disparition du caillot et cicatrisation des vaisseaux

5
Syndrome hémorragique : défaillance de l’hémostase physiologique
Hémostase : processus localisé et rapide, en équilibre physiologique entre processus coagulant et fibrinolyse, régulés eux-mêmes par des inhibiteurs et activateurs Plaquettes et facteurs de la coagulation Activateurs du plasminogène Hémostase primaire Coagulation Fibrinolyse Inhibiteurs physiologiques de la coagulation Inhibiteurs physiologiques de la fibrinolyse Syndrome hémorragique : défaillance de l’hémostase physiologique Thrombose ou CIVD : aberration de l’hémostase physiologique

6
Hémostase primaire Objectif : formation d’un clou plaquettaire ou thrombus blanc Suffisant pour arrêter l’hémorragie au niveau des petits vaisseaux Insuffisant mais indispensable pour les plus gros vaisseaux

7
Hémostase primaire Intervenants : Plaquettes
Paroi vasculaire (endothélium, sous endothélium) Fibrinogène vWf Conditions hémodynamiques (forces de cisaillement, fonction du calibre des Vx, vitesse d’écoulement)
 Fibrinogène. vWf. Conditions hémodynamiques (forces de cisaillement, fonction du calibre des Vx, vitesse d’écoulement)")
8
Hémostase primaire 2 phases successives : Temps vasculaire
Temps plaquettaire

9
Temps vasculaire Acteur principal : endothelium
Barrière de perméabilité sélective hémocompatible ( protéoglycanes) Surface > 6 500m² dont 80% dans la microcirculation Synthèse et sécrétion : vWF(corps de Weibel Palade), tPA, PAI Transformation des phospholipides membranaires (PG I2) Contrôle du tonus vasculaire Balance homéostasique entre Propriétés antithrombotiques : PG I2, NO, tPA, ADPase, GAG Propriétés prothrombotiques : vWF, PAI Equilibre de la thrombolyse physiologique : PAI, tPA
 Surface > 6 500m² dont 80% dans la microcirculation. Synthèse et sécrétion : vWF(corps de Weibel Palade), tPA, PAI. Transformation des phospholipides membranaires (PG I2) Contrôle du tonus vasculaire. Balance homéostasique entre. Propriétés antithrombotiques : PG I2, NO, tPA, ADPase, GAG. Propriétés prothrombotiques : vWF, PAI. Equilibre de la thrombolyse physiologique : PAI, tPA.")
10
Temps vasculaire Vasoconstriction immédiate des vaisseaux lésés
Petits vaisseaux uniquement Passive (paroi élastique) Puis active par contraction réflexe Prolongée et accrue par les substances libérées par les plaquettes : adrénaline, TX A2 ↓ du diamètre du vaisseau Ralentissement du débit sanguin favorisant interaction plaquettes/paroi lésée
 Puis active par contraction réflexe. Prolongée et accrue par les substances libérées par les plaquettes : adrénaline, TX A2. ↓ du diamètre du vaisseau. Ralentissement du débit sanguin favorisant interaction plaquettes/paroi lésée.")
11
Temps plaquettaire Adhésion Activation Agrégation

12
Plaquettes Cellules anuclées issues des MK Durée de vie : 7-8 j
Membrane : glycocallix, bicouche lipidique Contenu Granules denses : ADP, Ca2+, Sérotonine Granules α : vWf, fibrinogène, fibronectine, thrombospondine Facteur de croissance PDGF Protéines de coag = V, VIII, XI, XIII, PS, K Protéines de mb : GPIb-IX, GPIIb-IIIa

13
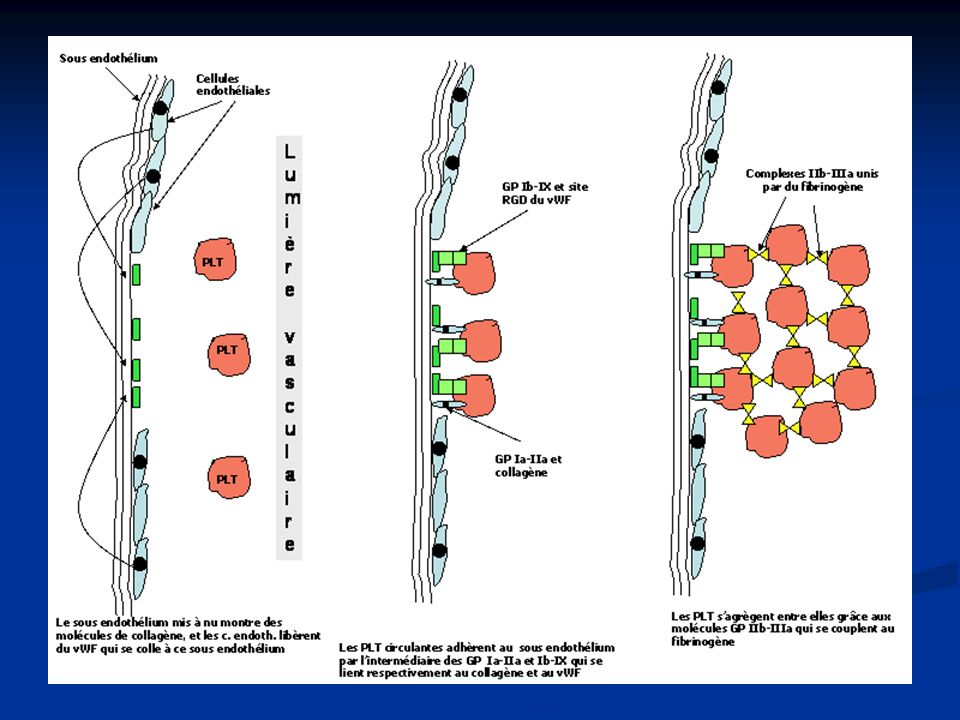
Temps plaquettaire Adhésion
Sous endothélium : thrombogène, composé de macromolécules (collagène, fibronectine, héparane sulfate) Plaquette/vWf Liaison du vWf au collagène Reconnaît son récepteur plaquettaire : GPIb-IX Modification conformationelle du vWf Plaquette/collagène Adhésion GPIb-IX indépendant (GPIa-IIa et GPIV)
 Plaquette/vWf. Liaison du vWf au collagène. Reconnaît son récepteur plaquettaire : GPIb-IX. Modification conformationelle du vWf. Plaquette/collagène. Adhésion GPIb-IX indépendant. (GPIa-IIa et GPIV)")
14
Temps plaquettaire Activation
Changement de forme et de structure interne Activation métabolique

15
Changement de forme et de structure interne
Temps plaquettaire Changement de forme et de structure interne Changement de forme Discoïde sphérique et volumineuse Etalement et émission de pseudopodes Libération des granules (release plaquettaire) Recrutement de plaquettes (ADP,…) Facteurs de croissance : modulation de la prolifération endothéliale Amplification de l’adhésion (vWf,…) Expression du GPIIb-IIIa (fusion membranaire) Activation du GPIIb-IIa (peut fixer ses ligands) Activité pro coagulante Flip flop = expression du PF3 Sert de support physique à la coagulation
 Recrutement de plaquettes (ADP,…) Facteurs de croissance : modulation de la prolifération endothéliale. Amplification de l’adhésion (vWf,…) Expression du GPIIb-IIIa (fusion membranaire) Activation du GPIIb-IIa (peut fixer ses ligands) 4. Activité pro coagulante. Flip flop = expression du PF3. Sert de support physique à la coagulation.")
16
Activation métabolique
Temps plaquettaire Activation métabolique Transformation des phospholipides membranaires : production de prostaglandines à partir de l’acide arachidonique PL membranaires Phospholipase Cyclo-oxygénase Thromboxane synthétase Phospholipase Cyclo-oxygénase Prostacycline synthétase Ac. arachidonique Composés intermédiaires PG I2 TX A2 Plaquette Cellule endothéliale AMPc ↓ - Agrégation +

17
Temps plaquettaire Agrégation plaquettaire
Processus actif nécessitant Ca2+, fibrinogène et de l’énergie En réponse à des stimuli : ADP, dérivés de l’acide arachidonique,… Fibrinogène fixé au GPIIb-IIIa sert de pont entre 2 plaquettes : réversible Thrombospondine et fibronectine : amplification et consolidation Clou plaquettaire Complété par la coagulation

19
EXPLORATION DE L’HEMOSTASE PRIMAIRE TEMPS DE SAIGNEMENT

20
Définition TS = temps nécessaire à l’arrêt du saignement d’une petite coupure (Vx de petite taille) Exploration in vivo des fonctions plaquettaires et vasculaires

21
Quand Indications : ATCD personnel et/ou familiaux de saignement (gingivorragie, epistaxis, ecchymose spontanée ou provoquée,…) Pathologie associée à des perturbations de l’hémostase primaire (IHC, IRC,…) PAS systématique dans le bilan pré-opératoire (l’interrogatoire est plus sensible)
 PAS systématique dans le bilan pré-opératoire. (l’interrogatoire est plus sensible)")
22
Comment TS = test global in vivo 3 techniques : DUKE
Incision au lobe de l’oreille Abandonnée (peu reproductible) IVY 3 points Standardisé Appareil à tension = Pression cste à 40 mmHg 3 points de piqûres face antéro-médiale de l’avant bras (zone sans Vx apparents) Intervalle et profondeur standardisés (2cm/2,5mm) Sang recueilli sur buvard / 30 sec sans toucher la brèche vasculaire Nle varie de 1 à 4 min Variation = sexe, âge, anémie Difficile à réaliser chez les personnes âgées (sang diffuse dans l’espace sous cutanée)
 IVY 3 points. Standardisé. Appareil à tension = Pression cste à 40 mmHg. 3 points de piqûres face antéro-médiale de l’avant bras (zone sans Vx apparents) Intervalle et profondeur standardisés (2cm/2,5mm) Sang recueilli sur buvard / 30 sec sans toucher la brèche vasculaire. Nle varie de 1 à 4 min. Variation = sexe, âge, anémie. Difficile à réaliser chez les personnes âgées (sang diffuse dans l’espace sous cutanée)")
23
Comment Techniques Ivy incision : référence
Incisions standardisées grâce à des dispositifs commerciaux : Surgicutt®, Simplate® Pression et recueil de sang identique Valeurs normales fonction du dispositif Nouveau-né : dispositif adapté

24
Comparaison Ivy 3 points et Ivy incision : sensibilité équivalente
Limites : Peu reproductible (opérateur dépendant) Standardisation nécessaire (Ivy incision> 3 pts) Peu sensible Pas prédictif du risque hémorragique Faux positifs : si incision trop profonde Atteinte des gros vaisseaux (coagulation) Rare cicatrice ( Ivy incision)
 Standardisation nécessaire (Ivy incision> 3 pts) Peu sensible. Pas prédictif du risque hémorragique. Faux positifs : si incision trop profonde. Atteinte des gros vaisseaux (coagulation) Rare cicatrice ( Ivy incision)")
25
Temps d’occlusion To (PFA-100)
Test in vitro Simule le passage du sang sur une brèche vasculaire Le sang citraté est déposé dans une cartouche test, puis aspiré dans un micro capillaire et traverse le micro orifice d’une membrane recouverte de collagène + EPI ou ADP Le système mesure le temps nécessaire à l’occlusion complète = TO Clou plaquettaire obstruant progressivement l’orifice

26
Temps d’occlusion To (PFA-100)
Avantages : Plus Se aux thrombopathies sévères (thrombasthénie de Glanzmann, Sd de J. Bernard et Soulier) Plus Se aux formes sévères de Willebrand (type 3, type 1 sévère) TO>TS pour la détection des Willebrand (?) Fait partie des tests de réponse au DDAVP Non opérateur dépendant Limites : N’explore pas les facteurs vasculaires Moins sensible pour les formes modérées de thrombopathies (granule), et Willebrand modéré
 Plus Se aux formes sévères de Willebrand (type 3, type 1 sévère) TO>TS pour la détection des Willebrand ( ) Fait partie des tests de réponse au DDAVP. Non opérateur dépendant. Limites : N’explore pas les facteurs vasculaires. Moins sensible pour les formes modérées de thrombopathies (granule), et Willebrand modéré.")
27
Conduite à tenir devant un allongement du TS
Eliminer une prise médicamenteuse Eliminer une erreur technique Incision trop ou pas assez profonde Buvard ne doit pas toucher l’incision Maintien d’une pression cste (brassard) Ininterprétable si Ht<25% Hb< 6g/dl Plq<70G/l Prélèvement>4h
 Ininterprétable si. Ht<25% Hb< 6g/dl. Plq<70G/l. Prélèvement>4h.")
28
Conduite à tenir devant un allongement du TS
Faire BC (TP, TCA, Fib)+ NFS-P
+ NFS-P.")
29
EXPLORATION DE L’HEMOSTASE PRIMAIRE PLAQUETTES

30
Plaquettes Anomalies quantitatives Thrombopénie (<50 G/L)
Anomalies qualitatives Thrombopathies acquises Thrombopathies congénitales (rare)
")
31
Thrombopénies

32
Thrombopénies Vrai thrombopénie ?
Frottis agrégats plaquettaires Prélèvement sur citrate Thrombopénie isolée ? (orientation diagnostic) GR : schizocytes, anisocytose, anémie,… GB : blastes, granulations anormales, … Plq : morphologie des plaquettes Origine centrale ou périphérique ? myélogramme
 GR : schizocytes, anisocytose, anémie,… GB : blastes, granulations anormales, … Plq : morphologie des plaquettes. Origine centrale ou périphérique myélogramme.")
33
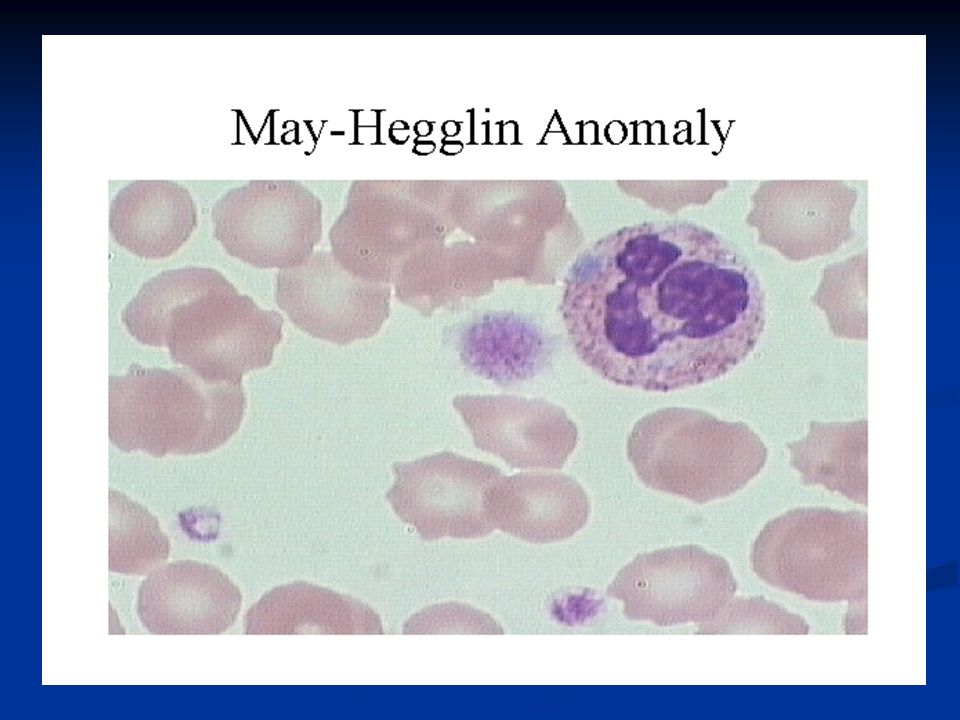
Thrombopénies Origines centrales : (pauvre en mégacaryocytes)
congénitale : ~ May Hegglin : plq géantes, inclusions basophiles dans les neutrophiles (corps de Döhle) ~ Wiskott Aldrich : petites plq, immunodépression, eczéma acquise : aplasie, hémopathies malignes, métastases, intox. alcoolique aiguë, benzène, médicaments (chimiothérapie, antiviraux, radiothérapie, …), carence en folate
 ~ Wiskott Aldrich : petites plq, immunodépression, eczéma. acquise : aplasie, hémopathies malignes, métastases, intox. alcoolique aiguë, benzène, médicaments. (chimiothérapie, antiviraux, radiothérapie, …), carence en. folate.")
35
Thrombopénies Origines périphériques (riche en mégacaryocytes)
Non immunologiques Dilution ~ Transfusions massives Consommation ~ CIVD +++ coag (fibrinogène, TP, TCA+++) ~ MAT (Purpura Thrombotique Thrombocytopénique , Syndrome Hémolytique et Urémique) ~ RM, valve mécanique, CEC, contre pulsion Par anomalie de répartition : ~ Hypersplénisme
 ~ MAT (Purpura Thrombotique Thrombocytopénique , Syndrome Hémolytique et Urémique) ~ RM, valve mécanique, CEC, contre pulsion. Par anomalie de répartition : ~ Hypersplénisme.")
36
Thrombopénies Origines périphériques Par destruction immunologique :
~ Purpura Thrombopénique Auto-Immun (PTAI) - infections (HIV++), MAI (lupus), hémopathies (LLC, Waldenström, lymphome) - Purpura Thrombopénique Idiopathique+++ - diagnostic d’élimination - 2/3 trombopénies ~ immuno-allergique : HEPARINE, quinine, digitaline, sulfamides, sels d’or, methyldopa… ~ allo-immunisation : thrompopénies néonatales (allo-Ac anti-PLT d’origine maternelle) et thrombopénies post- transfusionnelles
 - infections (HIV++), MAI (lupus), hémopathies. (LLC, Waldenström, lymphome) - Purpura Thrombopénique Idiopathique+++ - diagnostic d’élimination. - 2/3 trombopénies. ~ immuno-allergique : HEPARINE, quinine, digitaline, sulfamides, sels d’or, methyldopa… ~ allo-immunisation : thrompopénies néonatales (allo-Ac anti-PLT d’origine maternelle) et thrombopénies post- transfusionnelles.")
37
Thrombopathies

38
Thrombopathies Anomalies qualitatives fonctionnelles
Acquises : les + fréquentes - associées aux SMP, LA, Dysglobulinémies, IHC, IRC,… - médicamenteuses : Aspirine, Pénicilline, Ticlopidine,… Constitutionnelles : rares (1/10 Millions) Dystrophie de Bernard-Soulier : déficit GP Ib-IX (adhésion) Thrombasthénie de Glanzmann : déficit GP IIb-IIIa (agrégation) Maladie du pool vide : diminution contenu granules denses Syndrome des Plaquettes grises : diminution contenu granules α Déficit en Cyclo-oxygénase
 Dystrophie de Bernard-Soulier : déficit GP Ib-IX (adhésion) Thrombasthénie de Glanzmann : déficit GP IIb-IIIa (agrégation) Maladie du pool vide : diminution contenu granules denses. Syndrome des Plaquettes grises : diminution contenu granules α. Déficit en Cyclo-oxygénase.")
39
Diagnostic de thrombopathie

40
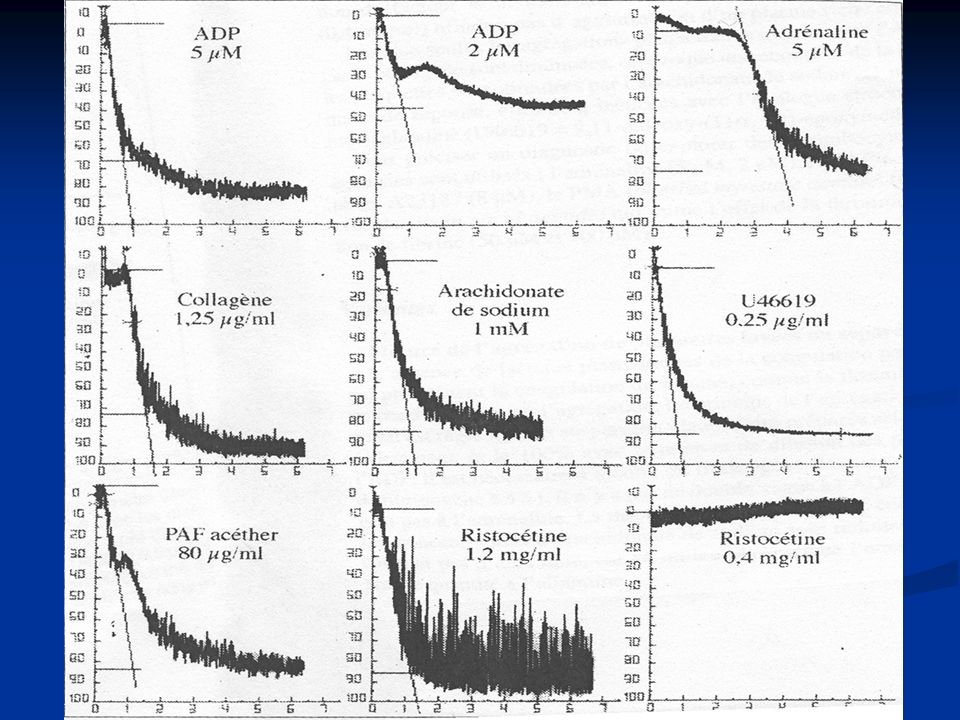
Agrégamétrie photométrique
Les plaquettes agrégent en présence d’agonistes Mise en évidence de l’agrégation : photométrie Lorsqu’on introduit l’agoniste, les plaquettes agrégent les unes aux autres ↓ particules en suspension et donc une ↑ rapide de la transmission lumineuse Profils d’agrégation plaquettaire (variation de transmission optique de PRP + différents agonistes) Ajout agoniste en 3 phases augmentation transmission optique (TO) ↓ TO (plq sphériques) Tps de latence puis ↑TO (agrégation) 3 paramètres Tps de latence (début agoniste/ début agrégation) % max d’agrégation Vélocité (pente de la courbe)
 Ajout agoniste en 3 phases. augmentation transmission optique (TO) ↓ TO (plq sphériques) Tps de latence puis ↑TO (agrégation) 3 paramètres. Tps de latence (début agoniste/ début agrégation) % max d’agrégation. Vélocité (pente de la courbe)")
42
Agrégamétrie photométrique
Agonistes de 1ere intention ADP Faible C : agrégation dpt fib/GP IIb IIIa Forte C : agrégation irréversible (dégranulation) Ristocétine Agrégation dpt GP Ib / vWf Faible C : pas d’agglutination Collagène Agrégation dépendant de la dégranulation et de la production de thromboxane A2
 Ristocétine. Agrégation dpt GP Ib / vWf. Faible C : pas d’agglutination. Collagène. Agrégation dépendant de la dégranulation et de la production de thromboxane A2.")
43
Agrégamétrie photométrique
Autres agonistes Ac. Arachidonique = suspicion de pathologie de la dégranulation ↓ agrégation avec ADP forte C et ↓avec Collagène Adrénaline, PMA,… (pathologie + rare, précise un diagnostic)
")
44
Principales anomalies des tests d’agrégation plaquettaire
Agoniste Glanzmann (GP IIb-IIIa) Bernard Soulier (GP Ib) Pseudo-Willebrand Déficit en granules denses Déficit granules a (PLT grises) ADP nulle Nle Diminuée et réversible Collagène Presque nulle Diminuée Ac. Arachi-donique Diminuée parfois Nle Ristocétine Présente mais diminuée Hypoagrég à faible dose Adrénaline
 Bernard Soulier. (GP Ib) Pseudo-Willebrand. Déficit en granules denses. Déficit granules a (PLT grises) ADP. nulle. Nle. Diminuée et réversible. Collagène. Presque nulle. Diminuée. Ac. Arachi-donique. Diminuée parfois Nle. Ristocétine. Présente mais diminuée. Hypoagrég à faible dose. Adrénaline.")
45
Réponses plaquettaires typiques à différents agents inducteurs en PRP

46
Autres tests Tests spécialisés orientés par l’agrégamétrie
Cytométrie en flux (étude des glycoprotéines) Métabolisme plaquettaire Etc..
 Métabolisme plaquettaire. Etc..")
47
EXPLORATION DE L’HEMOSTASE PRIMAIRE
Facteur Willebrand

48
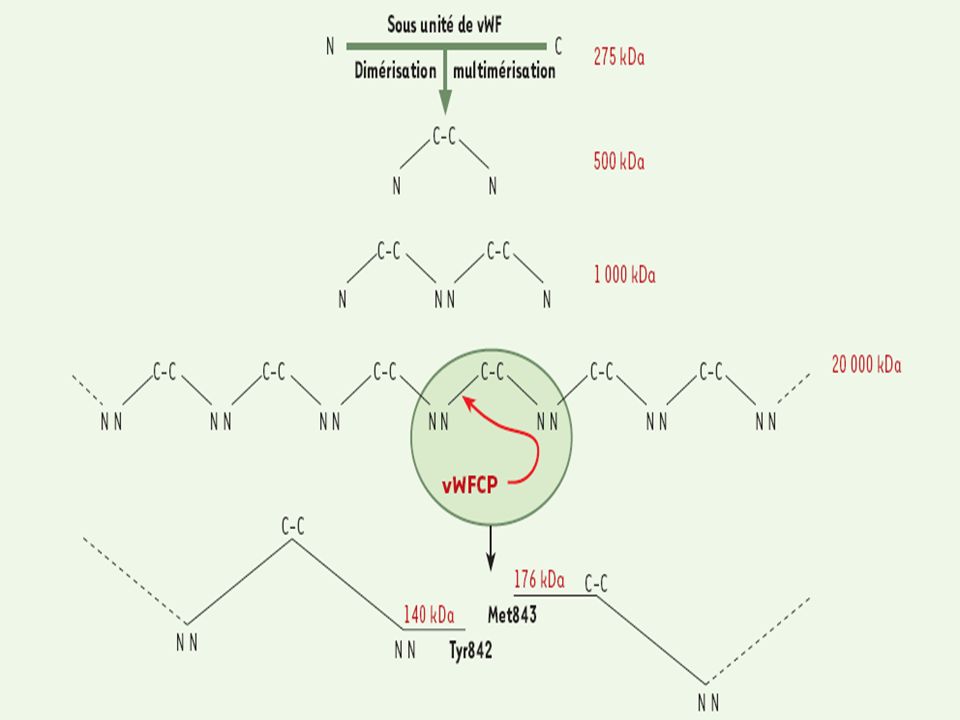
Willebrand - vWf Facteur de Willebrand :
Glycoprotéine synthétisée par les cellules endothéliales (corps de Weibel palade) et mégacaryocytes (granules) ∑ sous forme d’un précurseur (monomère) Actif sous forme de multimère (500 kDa = dimère kDa) Les multimères les plus longs sont les plus efficaces (+ gd nb de site d’interaction à ces ligands) De manière physiologique, une protéase, ADAMTS 13, coupe les sous-unités de manière aléatoire multimères + courts inhibe leur fixation spontanée aux plq Patho PTT, SHU (défaut action ADAMTS 13 cong ou acquis, -> multimères de très haut PM) microthrombi multiples Willebrand 2a (absence de multimères de HPM)
 et mégacaryocytes (granules) ∑ sous forme d’un précurseur (monomère) Actif sous forme de multimère (500 kDa = dimère kDa) Les multimères les plus longs sont les plus efficaces (+ gd nb de site d’interaction à ces ligands) De manière physiologique, une protéase, ADAMTS 13, coupe les sous-unités de manière aléatoire. multimères + courts. inhibe leur fixation spontanée aux plq. Patho. PTT, SHU (défaut action ADAMTS 13 cong ou acquis, -> multimères de très haut PM) microthrombi multiples. Willebrand 2a (absence de multimères de HPM)")
50
Willebrand - vWf 2 fonctions du vWF Transport et protection du VIIIc
Interactions plaquettes / paroi vasculaire lésée (adhésion) : liaison GP-Ib (ristocétine) et entre les plaquettes (agrégation) : GPIIb/IIIa
 : liaison GP-Ib (ristocétine) et entre les plaquettes (agrégation) : GPIIb/IIIa.")
51
Willebrand - vWf Variations physiologiques
Augmente au cours de la grossesse Des SRIS Ne pas dépister un Willebrand dans ces circonstances Taux plus faible chez les sujets de groupe sanguin O

52
Maladie de Willebrand Maladie hémorragique héréditaire la + fréquente
Autosomique dominante >> autosomique récessif 1 à 3% pop Déficit quantitatif et/ou qualitatif en vWf

53
Maladie de Willebrand Dépistage
Les tests globaux peuvent être réalisés TO>TS MAIS faible Se On peut d’emblée s’orienter vers des tests spécifiques vWf : RCO VIIIc +/- vWf Ag

54
Maladie de Willebrand Tests de dépistage TS augmenté
TCA souvent allongé (peut être Nle) Plaquettes souvent Nle (sauf IIb)
 Plaquettes souvent Nle (sauf IIb)")
55
Maladie de Willebrand Tests spécifiques de 1° intention - Buts
Diagnostic de Willebrand Typage : intérêt pronostique et thérapeutique vWf : RCO WIIIc +/- vWf Ag, si ↓ vWf : RCO (rapport vWf RCO/ vWf Ag)
")
56
Tests spécifiques Activité cofacteur de la ristocétine: vWf RCO
Agglutination de plaquettes Nle en présence du plasma du patient (vWf patient) et ristocétine (agrégamétrie) Ristocétine permet l’interaction vWf/plaquette (GPIb IX) Défaut d’agglutination = défaut quantitatif OU qualitatif de vWf Pb : la ristocétine n’est pas un agent physiologique Certains auteurs ont proposé d’utiliser du collagène pour apprécier l’aspect fonctionnel du vWf
 et ristocétine (agrégamétrie) Ristocétine permet l’interaction vWf/plaquette (GPIb IX) Défaut d’agglutination = défaut quantitatif OU qualitatif de vWf. Pb : la ristocétine n’est pas un agent physiologique. Certains auteurs ont proposé d’utiliser du collagène pour apprécier. l’aspect fonctionnel du vWf.")
57
Tests spécifiques Dosage du vWf antigène : vWf Ag
Quantité de vWf circulant Par ELISA Interprété en fonction du groupe sanguin Nle 50 – 200% Nle > 40% (groupe O)
")
58
Tests spécifiques Calcul du ratio vWf RCO/ vWf Ag
Ratio proche de 1 = anomalie quantitative Ratio < 0.7 = anomalie qualitative (activité du vWf trop basse par rapport au taux de vWf)
")
59
Tests spécifiques Mesure du VIIIc Normal ou abaissé
Responsable de la baisse du TCA

60
Tests spécifiques Autres tests pour différencier les anomalies
qualitatives: (sous types centres spécialisés) RIPA (ristocétine + PRP du patient) Analyse de la répartition des multimères Adhésion du vWf au VIII (type 2N)
 RIPA (ristocétine + PRP du patient) Analyse de la répartition des multimères. Adhésion du vWf au VIII (type 2N)")
61
Classification Type I 75% Iia 20% IIb 5% IIM multimère IIN normandie
Déficit partiel en vWF ↓ affinité du vWf pour plq ↑ affinité du vWf pour plq Anomalie fixation au VIII Déficit total (<1%) TO ↑ Nle ↑ ↑ ↑ VIIIc variable ↓ ↓ ↓ ↓ vW RCO N vWf Ag N ou ↓ vWRCO/vW Ag 1 <0.7 Multimère de haut PM + RIPA
 TO. ↑ Nle. ↑ ↑ ↑ VIIIc. variable. ↓ ↓ ↓ ↓ vW RCO. N. vWf Ag. N ou ↓ vWRCO/vW Ag. 1. <0.7. Multimère de haut PM. + RIPA.")
62
Test de réponse au DDAVP
DDAVP ↑ libération de vWf endothélial DDAVP CI dans forme IIb!!!!! Non indiqué dans la III et IIN Seul les sujets répondeurs pourront bénéficier de DDAVP Hgie mineure (pas de pronostic vital) « petite » chirurgie programmée Test indispensable (réponse individuelle variable)
 « petite » chirurgie programmée. Test indispensable (réponse individuelle variable)")
63
Test de réponse au DDAVP
Test : après administration de MINURIN® IV, OCTIM ® spray nasal TS ou TO VIIIc à HO, H1, H2, H4 vWf : RCO A H2 VIIIc et vWf RCO x 3 vWf > 30% Normalisation TO

64
Diagnostic différentiel
Hémophilie A modérée (2N++) Étude de la fixation du VIII au vWf Hémophile A sévère et type III Maladie de Willebrand acquise (Ig monoclonale, MAI,…) Pseudo Willebrand (anomalie du GPIb-IX)
 Étude de la fixation du VIII au vWf. Hémophile A sévère et type III. Maladie de Willebrand acquise (Ig monoclonale, MAI,…) Pseudo Willebrand (anomalie du GPIb-IX)")
65
EXPLORATION DE L’HEMOSTASE PRIMAIRE Fibrinogène

66
Fibrinogène Afibrinogénémie congénitale
Autosomique récessive, exceptionnelle Déficience complète en fibrinogène Fibrinogène < 0,2 g/l Diagnostic évoqué en période néonatale Hgie importante, pour des traumatismes minimes Possibilité d’Hgies intra crâniennes

67
EXPLORATION DE L’HEMOSTASE PRIMAIRE Paroi vasculaire

68
Paroi vasculaire Diagnostic d’élimination Pathologies
Maladie de Rendu Osler Anomalies du collagène (Ehlers Danlos,…) Sinon : « fragilité capillaire »…
 Sinon : « fragilité capillaire »…")
69
Conclusion Exploration de l’hémostase I:
Test de 1ère intention : TS/TO Seul test global de l’hémostase I MAIS grandes limites : Manque de Se +++ Sa normalité n’exclue pas une anomalie de l’hémostase I Tests de 2ème intention Peuvent être réalisés d’emblée si suspicion clinique forte Tests de 3ème intention Précisent le diagnostic

Présentations similaires








>")










