Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Oncogénétique Prédisposition au Cancer du Colon et au Mélanome Formes monogéniques, implications médicales

3
Oncogénétique: étude des cancers « héréditaires »
Quels sont les patients et les familles concernées? Quelles sont les modalités de prise en charge? Anomalies des cellules tumorales Observations épidémiologiques, formes monogéniques L’exemple du rétinoblastome Génétique de cancers familiaux Cancer colorectal Cancer du sein

4
Anomalies génétiques dans les cellules tumorales
Les cellules tumorales comportent des anomalies génétiques Il existe une accumulation progressive des mutations Certaines mutations sont sélectionnées (évolution clonale) activation d’oncogènes inactivation de gènes suppresseurs de tumeur
 activation d’oncogènes. inactivation de gènes suppresseurs de tumeur.")
5
Types d’altérations génétiques
Activation d’oncogènes = 1 seul évènement Mutation gain de fonction, ex c-kit dans GST Translocation, ex bcr-abl dans LMC Amplification, ex Cyclin D1 Inactivation de gènes suppresseurs de tumeur= 2 évènements CDKN2a (p16) P53
 P53.")
6
CYCLE CELLULAIRE cdc2 - Cycline B Cycline D M G1 G2 G0 Point START
cdk2 cdk4 Cycline D cdk5 S cdk6 cdk2 - Cycline E cdk2 - Cycline A

7
Prédisposition génétique au cancer
= Agrégation de cancers dans une même famille * Rare * Age plus précoce/ Cas sporadique * Plusieurs cancers chez un même individu Pénétrance: Forte/faible - Transmission Autosomique dominante ++++: Gènes suppresseurs de tumeur (ex: BRCA1, hMLH1, CDKN2A, p53) + rares cas oncogènes (CDK4) Récessive (Xeroderma pigmentosum) Polygénique
 + rares cas oncogènes (CDK4) Récessive (Xeroderma pigmentosum) Polygénique.")
8
- - - - - p16INK4a p14ARF + oncogènes Arrêt du Cycle apoptose p21 Bax
M G1 G2 Arrêt du Cycle apoptose Point START - S + p21 Bax E2F + RB-E2F gènes tumeur- suppresseurs p53 - Cdk4-cyclin D Cdk6 P - MDM2 dégradation oncogènes - - transcrit transcrit CDKN2A p16INK4a p14ARF 5

9
Evolution clonale mutation mutation mutation mutation Hypertrophie
Each cell, when it divides, generates two identical new ones. So, when a cell acquires a mutation, it passes that mutation on to its progeny during cell growth and division. Because cells with cancer-linked mutations tend to proliferate more than normal cells, cellular candidates for additional mutations grow in number. Mutations continue to accumulate and are copied to descendant cells. If one cell finally acquires enough mutations to become cancerous, subsequent cancer cells will be derived from that one single transformed cell. So all tumors are clonal, which means that they originate from a single parent cell, whether that first mutant cell was of germline or somatic origin. Hypertrophie épithéliale adénome carcinome métastases

10
Observations épidémiologiques
Etudes de population: risque relatif des apparentés augmenté (X 1,5- 3) Facteurs génétiques multiples+ environnement partagé implications individuelles modestes (Prévention, dépistage)
 Facteurs génétiques multiples+ environnement partagé. implications individuelles modestes. (Prévention, dépistage)")
11
Arbres généalogiques évocateurs d’une transmission mendélienne
Cas « rares »: 2-5% des cancers CCR 68 ans CCR 53 ans CCR 45 ans K utérus (53 ans) Implications individuelles majeures
 Implications individuelles majeures.")
12
L’exemple du rétinoblastome
Tumeur rare de la rétine chez l’enfant Cas sporadiques et cas familiaux Bilatéralité des cas familiaux

13
Hypothèse de Knudson (1971)
Deux anomalies génétiques sont nécessaires pour initier la formation d'un rétinoblastome. Dans les cas de rétinoblastomes héréditaires, une anomalie est héritée d’un parent La deuxième anomalie résulte d’une mutation somatique

14
Identification du gène Rb (1986)
Etudes des cancers familiaux Dans quelques familles, des délétions d'une région chromosomique 13q14 se transmettent de façon héréditaire avec le phénotype Perte d'allèles dans les tumeurs au niveau de la même région

15
pRb La protéine Rb E2F Cycline D - cdk4 P Prolifération cellulaire
Facteur de transcription Cycline D - cdk4 P Expression des gènes de la phase S + Prolifération cellulaire Cellule normale: progression du cycle dépend de la phosphorylation de Rb

16
+ pRb E2F Prolifération cellulaire
Inactivation de Rb prolifération non contrôlée pRb E2F Facteurs de transcription Expression des gènes de la phase S + Prolifération cellulaire

17
Transmission héréditaire
Mutation somatique Mutation germinale transmise Sporadique Premier mutation somatique Deuxième mutation somatique

18
Perte de chromosome et duplication Mutation ou methylation
Allele normal Allele muté Perte de l’allèle normal Perte de chromosome et duplication Perte de Chromosome Deletion Mutation ou methylation In hereditary cancer syndromes, individuals are called heterozygous (having one or more dissimilar gene pairs) because they start life with a germline mutation in one of the alleles linked to cancer susceptibility, but it is balanced by a normal counterpart. These individuals are predisposed to cancer because all their cells have already sustained the first hit to cancer-linked genes. If the critically needed normal suppressor gene that balances this germline mutation is lost at some time during an individual’s life, a condition called loss of heterozygosity (LOH) occurs.
 because they start life with a germline mutation in one of the alleles linked to cancer susceptibility, but it is balanced by a normal counterpart. These individuals are predisposed to cancer because all their cells have already sustained the first hit to cancer-linked genes. If the critically needed normal suppressor gene that balances this germline mutation is lost at some time during an individual’s life, a condition called loss of heterozygosity (LOH) occurs.")
19
Le deuxième évènement ne se produit pas toujours:
pénétrance incomplète

20
En pratique: consultation d’oncogénétique
Quels sont les patients et les familles concernées? Formes « monogéniques » de prédisposition aux tumeurs: Risque élevé pour les individus génétiquement prédisposés Possibilité de prévention Possibilité de diagnostic anténatal

21
Le conseil génétique Analyse précise et détaillée de l’histoire familiale Causes de décès et maladies des membres de la famille Age au moment du diagnostic Demande de complément d’information (CR anatomopathologiques) Évaluer la probabilité de l’existence d’une forme monogénique
 Évaluer la probabilité de l’existence d’une forme monogénique.")
22
Conseil génétique (2) - Confirmer le diagnostic (Formes syndromiques)
Organiser le conseil génétique * Test moléculaire : cas index * Diagnostic présymptomatique : apparentés (Mélanome familial, Syndrome de Birt Hogg Dube) * Diagnostic anténatal : si morbidité importante (Xeroderma pigmentosum) - Mesures de prévention
 * Diagnostic anténatal : si morbidité importante (Xeroderma pigmentosum) - Mesures de prévention.")
23
1ère consultation d’oncogénétique
Par un médecin généticien Proposée, non imposée, consultation longue En collaboration avec le staff de cancérologie Information du patient : possibilités diagnostiques et de prévention Arbre généalogique avec recueil de tous les cas de cancers Prélèvement génétique + signature d’un consentement éclairé attestation aide psychologique si besoin

24
Consentement éclairé, prescription du test
(loi du 6 août 2004 modifiant la loi du 29 Juillet 1994) Démarche de génétique recueil d’un consentement de cette personne par écrit médecin oeuvrant au sein d’une équipe pluridisciplinaire agrément des laboratoires et des praticiens pour une période de 5 ans. Confidentialité Devoir d’information Autonomie du sujet Informations sur la maladie Nature du test génétique Implications du résultat + ou -
 Démarche de génétique. recueil d’un consentement de cette personne par écrit. médecin oeuvrant au sein d’une équipe pluridisciplinaire. agrément des laboratoires et des praticiens pour une période de 5 ans. Confidentialité. Devoir d’information. Autonomie du sujet. Informations sur la maladie. Nature du test génétique. Implications du résultat + ou -")
25
d’intérêt 2 Graphisme : iconographie, Institut Curie

26
ANALYSE AU LABORATOIRE
Etude du cas index SEQUENCE CODANTE DES GENES Mutation identifiée NON Recherche des grands réarrangements, analyse des promoteurs OUI Mutation identifiée NON Confirme le diagnostic Permet l’étude des apparentés N’exclut pas le diagnostic

27
Deuxième consultation
Rendu du résultat du test au patient Explication : conséquences en matière de prévention et de suivi au sein de la famille Proposition de diagnostic présymptomatique

28
Analyse moléculaire chez un sujet asymptomatique = diagnostic présymptomatique
Arrêté du 02 mai 2001 : Dans le cadre d’une équipe pluridisciplinaire Compétence en génétique Consultation individuelle Mutation familiale connue 1 ou 2 consultations avant prélèvement sanguin

29
Exploration moléculaire
chez un apparenté, Recherche de la mutation identifiée chez le cas index Par séquençage de l’exon porteur de la mutation

30
Résultat rendu lors d’une consultation
Absence de mutation : Pas de risque familial Risque général persiste Pas de transmission du risque à la descendance Mutation présente : Risque liée à la prédisposition familiale Prise en charge: prévention Risque de transmission

31
Rôles du médecin traitant ( du dermatologue)
Dépiste les formes évocatrices Participe au diagnostic des formes génétiques Prend en charge le traitement du patient

32
Rôles de l’onco-généticien
Précise le risque Test génétique du cas index En cas de mutation > organise le suivi avec équipe pluridisciplinaire > organise le test des apparentés

33
Prédisposition héréditaire au cancer colo-rectal (CCR)
Fréquence cancer du colon: 1/200 95% tumeurs sporadiques 5 à 10% des CRC prédisposition familiale HNPCC (1% des CCR) Polypose adénomateuse familiale
 Polypose adénomateuse familiale.")
34
POLYPOSE COLIQUE FAMILIALE
Transmission autosomique dominante Age moyen 39 ans, multiples polypes du colon +/- intestin grêle et estomac Cancers associés : intestin grêle, estomac, thyroïde Mutations du gène APC (chromosome 5q) Traitement : colectomie totale ans
 Traitement : colectomie totale ans.")
38
Syndrome de LYNCH ou HNPCC (Hereditary Non Polyposis
Colorectal Cancer)
")
39
Systèmes de réparation de l’ADN
Mutations de l’ADN Mutagènes Erreurs de Replication Systèmes de réparation de l’ADN SRM = Système de réparation des Mésappariements = couplé à l’ADN polymérase répare les incorporations de bases fautives dans les séquences répétées

40
SRM suite Complexe multiprotéique hMSH2, hMLH1, PMS1, PMS2, hMSH6
Altération du SRM: Taux de mutations dans la Cellule tumorale Vitesse d’accumulation taux mutations dans oncogènes et gènes suppresseurs SRM +/- pas d’altération de la réparation SRM -/- Instabilité des séquences microsatellites, accumulation mutations ponctuelles dans les gènes

41
Correction des erreurs de réplication
replication AT NNNATATAT ATATAT NNNTATATA TATATATATATANNN insertion SRM NNNATATATATATATATATATNNN NNNTATATATATATATATATANNN SRM NNNATATAT ATATAT NNNTATATA TATATATATATANNN TA deletion

42
Instabilité des séquences simples répétées (microsatellites)
TATATATATATATATATA ADN Analyse des Produits de PCR Phénotype tumoral MSI Sang tumeur

43
SYNDROME HNPCC (1) (cancer du colon héréditaire sans polypose) = Hereditary Non Polyposis Cancer
Suspicion d’un caractère héréditaire depuis le début du siècle Mais : Hétérogénéité génétique, pénétrance incomplète, phénocopies Difficulté de clonage des gènes Etapes du clonage: Liaison génétique: 2 loci 2p16 et 3p21 Instabilité microsatellite des tumeurs Clonage chez E Coli des gènes MutH, MutL, MutS Clonage chez l’homme des homologues (hMSH2, hMSH1) Confirmation de leur rôle dans l’HNPCC: Mutation germinale Correction du phénotype mutateur des tumeurs hMLH1 -/- par surexpression hMLH1
 Confirmation de leur rôle dans l’HNPCC: Mutation germinale. Correction du phénotype mutateur des tumeurs hMLH1 -/- par surexpression hMLH1.")
44
Syndrome HNPCC (2): Génétique
• Transmission AD pénétrance incomplète 50-80% Fréquence hétérozygotes 1/500 Age moyen 44 ans Cancers associés ++ : risque élevé endomètre- rein-uretère risque plus faible intestin grêle-estomac- ovaire Tumeurs : phénotype RER (instabilité microsatellites) Mutations germinales de gènes impliqués dans la correction des misappariements (mismatch) de l’ADN : système RM (réparation des mismatchs)
 Mutations germinales de gènes impliqués dans la correction des. misappariements (mismatch) de l’ADN : système RM (réparation des mismatchs)")
45
Syndrome HNPCC SPECTRE TUMORAL Spectre tumoral étroit:
Cancer colo-rectal (colon droit, peu différencié, composante mucineuse, instabilité micro-satellitaire) Cancer de l’endomètre Cancer intestin grèle Carcinome urothélial voies urinaires supérieures Spectre tumoral élargi: Adénocarcinome gastrique Cholangio-carcinome Cancer de l ’ovaire (ADK endométrioïde) Glioblastome (syndrome de Turcot) Carcinome sébacé (Syndrome de Torre-Muir)
 Cancer de l’endomètre. Cancer intestin grèle. Carcinome urothélial voies urinaires supérieures. Spectre tumoral élargi: Adénocarcinome gastrique. Cholangio-carcinome. Cancer de l ’ovaire (ADK endométrioïde) Glioblastome (syndrome de Turcot) Carcinome sébacé (Syndrome de Torre-Muir)")
46
Cinq gènes différents 2p15 hMSH2 (homologue de MutS) 45%
3p21 hMLH1 (homologue de MutL) 49% 7p22 hPMS2 (homologue de MutL) 6% 2q31 hPMS1 (homologue de MutL) rares 2p16 hMSH6 (homologue de MutS) rares x x Progression tumorale Inactivation du 2ème allèle: Instabilité génétique (SRM-) Mutation germinale: Génome stable (SRM+)
 49% 7p22 hPMS2 (homologue de MutL) 6% 2q31 hPMS1 (homologue de MutL) rares. 2p16 hMSH6 (homologue de MutS) rares. x. x. Progression tumorale. Inactivation du 2ème allèle: Instabilité génétique (SRM-) Mutation germinale: Génome stable (SRM+)")
47
MutS

48
Critères cliniques faisant suspecter un Syndrome HNPCC
= indication à une consultation oncogénétique 1) Risque élevé (critères Amsterdam II): sensibilité 60-70% - Au moins 2 cas de cancer familial évocateur (CCR, endomètre, estomac grêle, rein, voies biliaires) dont au moins deux au premier degré - CCR sur au moins 2 générations - Age au Diagnostic ≤ 50 ans chez un des malades 2) Risque intermédiaire - CCR ou Cancer du spectre HNPCC < 40 ans - CCRs multiples ou CCR + K endomètre, CCR + autre K spectre HNPCC - Tumeur MSI + < 60 ans, cancer colique gauche ou rectal MSI + • Nombre prévisionnel de cs d’oncogénétique = 5 à 10% des cas incidents, Soit 2000/ an
 Risque élevé (critères Amsterdam II): sensibilité 60-70% - Au moins 2 cas de cancer familial évocateur (CCR, endomètre, estomac. grêle, rein, voies biliaires) dont au moins deux au premier degré. - CCR sur au moins 2 générations. - Age au Diagnostic ≤ 50 ans chez un des malades. 2) Risque intermédiaire. - CCR ou Cancer du spectre HNPCC < 40 ans. - CCRs multiples ou CCR + K endomètre, CCR + autre K spectre HNPCC. - Tumeur MSI + < 60 ans, cancer colique gauche ou rectal MSI + • Nombre prévisionnel de cs d’oncogénétique = 5 à 10% des cas incidents, Soit 2000/ an.")
49
- Famille HNPCC ? K utérus CCR 45 ans Jean CCR 53 ans CCR 68 ans 2 mm

50
Eléments en faveurs HNPCC
3 cas de CCR Transmission AD avec pénétrance incomplète Age jeune Cancer apparenté HNPCC

51
Etapes de démarche diagnostique
Analyse de l’ADN tumoral/ADN leucocytaire Présence d’un phénotype MSI + 2) Si MSI +: recherche de mutations dans hMSH2, hMLH1 3) Si présence de mutations: recherche chez les apparentés (majeurs) pour diagnostic présymptomatique
 Si MSI +: recherche de mutations dans hMSH2, hMLH1. 3) Si présence de mutations: recherche chez les apparentés (majeurs) pour diagnostic présymptomatique.")
52
Diagnostic indirect Mutation génétique Marqueurs Protéine
ADN leucocytaire hétérozygote allèles Présente ADN tumoral homozygote n allèles Absente 0% activité résiduelle x 50% activité résiduelle x Mutation germinale 2ème événement somatique

53
Recherche du phénotype MSI
Procédure standardisée par le NIH Génotypage de 5 marqueurs monomorphes: (BAT25, BAT26, NR21, NR24, NR27) sensibilité >10% d’ADN tumoral
 sensibilité >10% d’ADN tumoral.")
54
Immunohistochimie Anticorps anti-hMLH1, anti-hMSH2, anti-hMSH6
Perte d’expression dans la tumeur/ muqueuse normale sensibilité < génotypage (92%) Importantes variations d’interprétations inter-observateurs indication sur la mutation causale (hMSH2 +++)
 Importantes variations d’interprétations inter-observateurs. indication sur la mutation causale (hMSH2 +++)")
55
Recherche de mutations germinales
Recherche de mutation ponctuelles hMLH1 et hMSH2 Puis séquençage de hMSH6 Recherche de réarrangement de grande taille si forte suspicion

56
Conseil génétique: implications familiales
* Mutation chez le cas index Recherche de mutation chez les apparentés Si mutation: mise en place d’un dispositif: • Coloscopie dés 25 ans puis tous les deux ans, à compléter par un colorant de type rouge carmin • Chirurgie prophylactique= pas d’indication - Si pas de mutation: on rassure l’individu * Pas de mutation chez le cas index: on ne peut pas exclure HNPCC: Surveillance coloscopique de la famille

57
Prise en charge gynécologique
Oestroprogestatifs non Cind Examen gynécologique annuel >30 ans * Mesure de l’épaisseur endomètre par échographie * Hystéroscopie En cours d’évaluation - Hystérectomie prophylactique ds certains cas (CCR)
")
58
Conseil génétique - Consultation d’oncogénétique Suspiscion HNPCC +
Phénotype MSI Cancer du colon + Diagnostic moléculaire MLH1, MSH2 Test des apparentés + - Surveillance ciblée Surveillance de tous les apparentés 44

59
Melanoma risk factors Genes (OMIM 155600) Other risk factors
Phenotypic/host factors Rare mutations conferring high risk -2% Nevus: number, dysplasia Pigmentation phenotype fair skin, freckles, clear eyes, blond or red hair Skin reaction to sun:Phototype I-II inability to tan, propensity to sunburn CDKN2A Melanocyte CDK4 BAP1 [Nevi] Frequent variants conferring low risk Environmental factor MC1R , OCA2, ASIP, TYR, MATP, MITF, ATM… • UV/sun exposure Melanoma

60
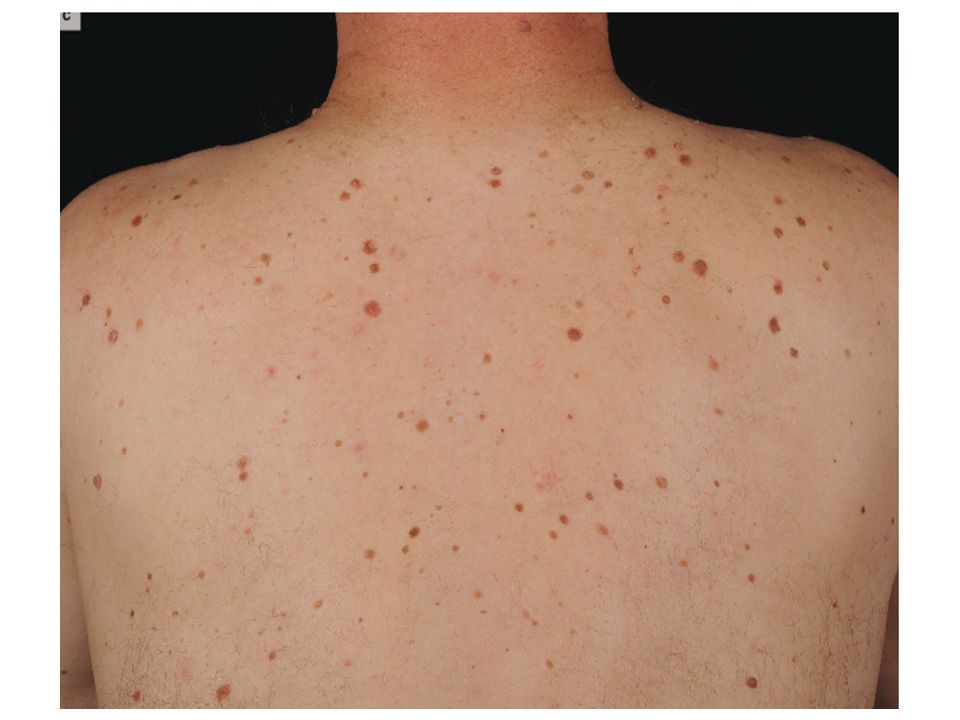
Mélanome Familial • 5 à 10% • Clinique - Jeune âge au diagnostic
- Syndrome naevus atypique 50% - Mélanome Multiple 12 à 40% - Cheveux Roux 2

61
Facteurs de risque génétiques (1) Gènes de prédisposition majeurs
• autosomique dominante • forte pénétrance 40-60% • 3 gènes susceptibilité CDKN2A en 9p21 (1994) CDK4 en 12q14 (1996) BAP1 (octobre 2011) • autres loci 1p36, 1p22, 9q31… 3
 CDK4 en 12q14 (1996) BAP1 (octobre 2011) • autres loci 1p36, 1p22, 9q31… 3.")
62
Tumor suppressor genes
apoptosis START Point - S + p21 Bax E2F + RB-E2F Tumor suppressor genes p53 - MDM2 dégradation Cdk4-cyclin D Cdk6 P oncogenes - - transcrit transcrit CDKN2A p16INK4a p14ARF 5

63
- * * * * Mutation CDKN2A Ala68Leu Cancer pancréas 2 5 mélanomes
2 mm - * 20

64
Mutation Thr77Pro Leu-33 P16INK4A Arg-31 CDK4

65
Mutations de CDKN2A et mélanome familial
Familles mutées % Suède sur Australie 22 sur USA sur UK sur Canada 23 sur Espagne sur Italie sur Pays Bas sur • % • Ségrégation • Impact fonctionnel 16

66
Implication de CDKN2A (2)
• Cancer du pancréas Risque x 13 dans familles mutées Cancer du pancréas familial • Mélanome multiple sporadique Mutation germinale 6 à 10 % des cas • Cancer épidermoïde, K sein, myelome ? 17

67
Tumoral risk of CDKN2A carriers within melanoma-prone families
Melanoma depends on (1) geographic location : 58% by age 80 in Europe, 76% in USA, 91% in Australia (UV) (2) modifier genes: MC1R genotype, …. (3) other associated risk factors (naevi count, phototype, UV) Pancreatic cancer risk of 15% by age 80: family dependant, tabagism, mutation dependant : Leiden, Gly101Trp ++ Others cancers NST, breast, : controversial Bishop TD et al, JNCI, 2002
 geographic location : 58% by age 80 in Europe, 76% in USA, 91% in Australia (UV) (2) modifier genes: MC1R genotype, …. (3) other associated risk factors (naevi count, phototype, UV) Pancreatic cancer risk of 15% by age 80: family dependant, tabagism, mutation dependant : Leiden, Gly101Trp ++ Others cancers NST, breast, : controversial. Bishop TD et al, JNCI,")
68
Cancer du pancréas et mutation de CDKN2A
Cancer du pancréas familial 3.3% Cancer pancréas + apparenté mélanome 5.5% Pénétrance 58% à 80 ans HR 25.8 chez les fumeurs mutés / non fumeurs mutés

69
Gène CDK4 et mélanome familial
2 mutations germinales toutes situées dans l ’exon 2 Mutations activatrices: oncogène Littérature: 13 familles < 2 % CDK4 - Second gène de prédisposition - Moins impliqué que CDKN2A 23

70
Oncogénétique : Mélanome familial
Quels sont les patients et les familles concernées ? Quand adresser un patient en consultation d’oncogénétique ? Quelles sont les modalités de prise en charge et la législation?

71
2 5 mélanomes 2 mélanomes 2 mélanomes 2 mm - 20

72
Test CDKN2A/CDK4 chez les mineurs? 01/2011
Etant donné que Les mélanomes sont très rares chez les mineurs dans les familles p16+ L’impact psychologique d’un « étiquetage » muté dans une fratrie Qu’il n’y a pas de bénéfice direct car la photoprotection et la surveillance (en vue d’un dépistage précoce) doit être la même chez tous les enfants Que le risque est grand de perdre la compliance chez les adolescents P16- Qu’il est important de laisser le libre choix à la personne Il est décidé à l’unanimité de ne pas recommander les tests chez les mineurs, malgré la pression parentale et des opinions divergentes exprimées dans la littérature (Taber JM, Genet Med, 2010)
 doit être la même chez tous les enfants Que le risque est grand de perdre la compliance chez les adolescents P16- Qu’il est important de laisser le libre choix à la personne. Il est décidé à l’unanimité de ne pas recommander les tests chez les mineurs, malgré la pression parentale et des opinions divergentes exprimées dans la littérature (Taber JM, Genet Med, 2010)")
73
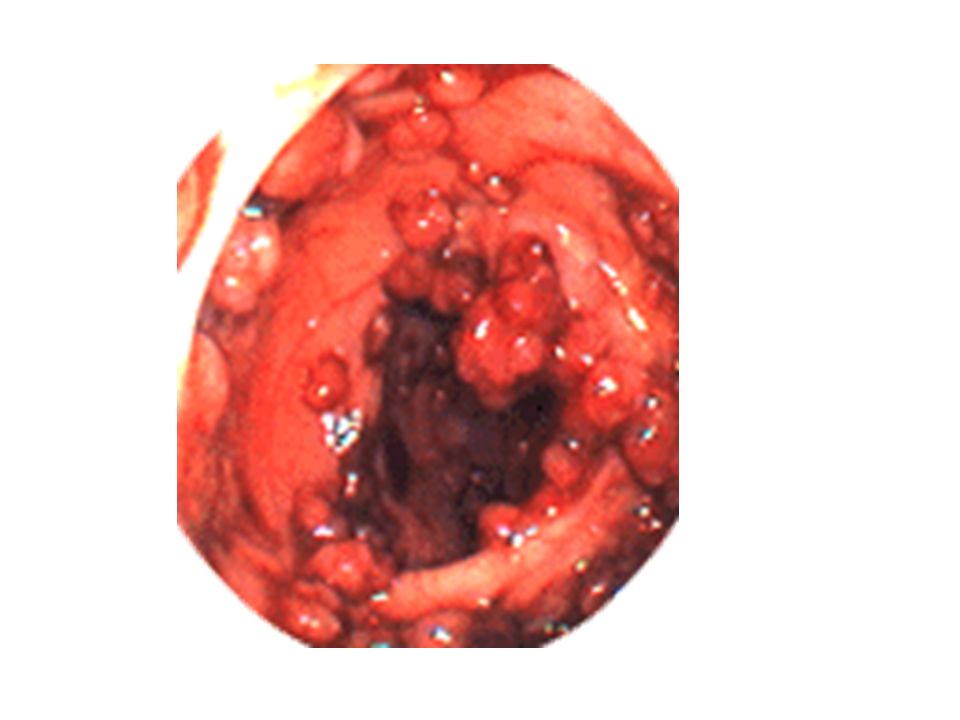
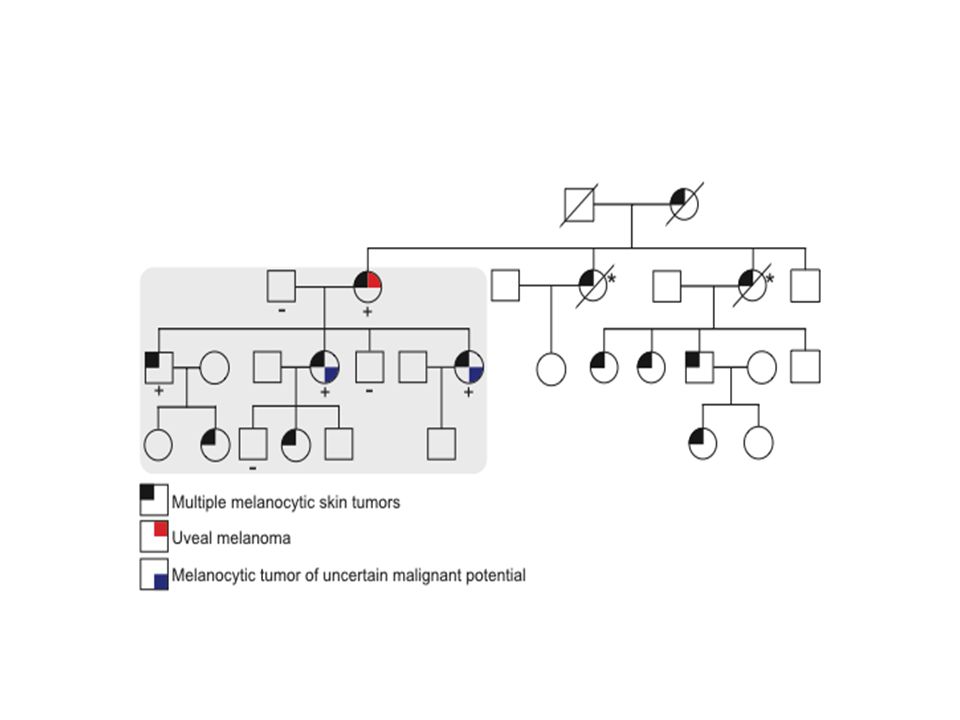
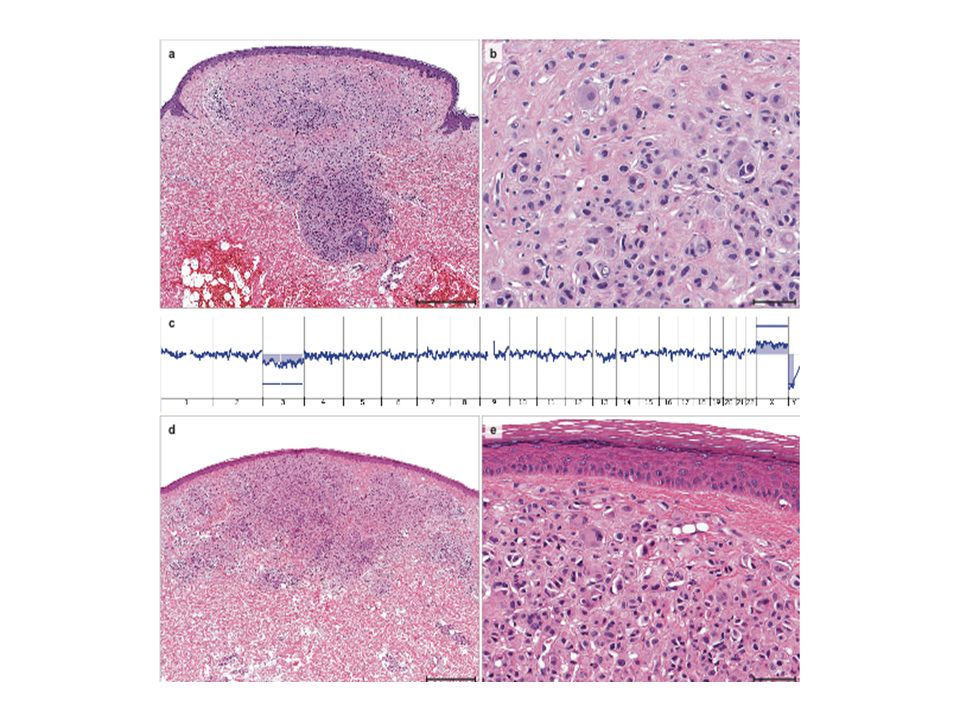
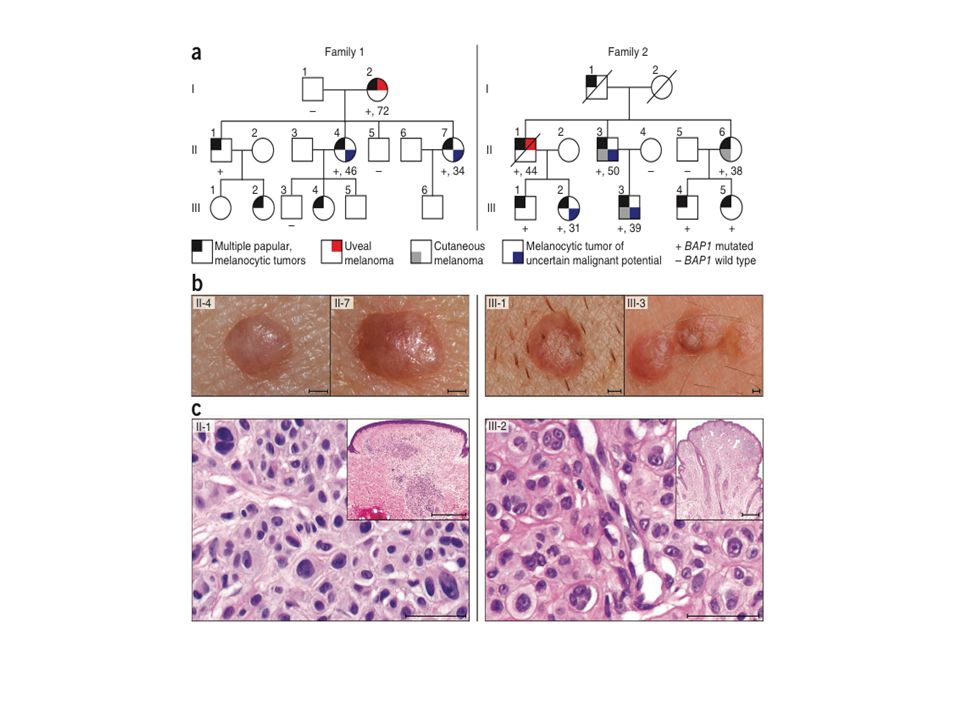
Wiesner et al, Nature Genetics, Octobre 2011
2 familles Nevus dermiques couleur peau > brun orangés de 5 mm Histologie particulière : large vésicules nucléaires de taille inégale « spitzoïde » Tumeurs mélanocytaires de malignité incertaine Mélanome choroïde++ Mélanome cutané

79
Identification de mutations constitutionnelles de BAP1
CGH arrays NGS 3p21

80
Tumeurs mélanocytaires atypiques
Perte d’expression de BAP1 2 contingents de cellules

81
Quel est le rôle de BAP1 dans l’oncogenèse du mélanome ?
156 mélanomes Séquençage BAP1 Mutations BAP1 dans - 40% UMM - 11% des naevus de Spitz atypiques - 5% des mélanomes cutanés

82
La transformation du nevus en mélanome est associée à une perte de BAP1
80% des tumeurs mélanocytaires mutées BRAF Perte du 2ème allèle par délétion ou mutation ponctuelle

83
BAP1 BAP1= partenaire de BRCA1 Enzyme déubiquinante Liaison a HCF1
G1/S transition Impliqué dans la réponse aux dommages de l’ADN, le cycle cellulaire, l’apoptose, la sénescence Prédispose aussi au mésothéliome ++

84
Et notre expérience ? 15 familles de mélanomes dont au moins un apparenté atteint de UM Une nouvelle mutation détectée

85
c.588 G>A hétero, p.Trp196Ter
DCD 89 ans Infarctus DCD 72 ans DCD 70 ans 2 grands oncles/tantes pat+ 1 petite cousine pat atteints K colon 1933 1939 Cancer prostate en 2002 Mélanome oculaire 69 ans DCD 71 ans 1959 SSM Mollet D, Clark I CDKN2A wt CDK4 wt BAP1 c.588 G>A hétero, p.Trp196Ter 1964 1963 1988 1964 1964

86
Mutations somatiques de BAP1 et mélanome oculaire
LOH 3p21

87
Indications du test génétique
1) Mélanome Cutané Au moins 2 cas de mélanomes invasifs sur la même branche parentale Au moins 2 mélanomes invasifs chez la même personne Un cancer du pancréas peut remplacer un mélanome 2) Mélanome Oculaire Mélanome de la choroïde familial associé à mélanome cutané dans la famille associé à mésothéliome dans la famille Seuil de 10% de détection de mutation
 Mélanome Cutané. Au moins 2 cas de mélanomes invasifs sur la même branche parentale. Au moins 2 mélanomes invasifs chez la même personne. Un cancer du pancréas peut remplacer un mélanome. 2) Mélanome Oculaire. Mélanome de la choroïde familial. associé à mélanome cutané dans la famille. associé à mésothéliome dans la famille. Seuil de 10% de détection de mutation.")
88
Conseil génétique Consultation d’oncogénétique Mélanome héréditaire
Test des apparentés Diagnostic moléculaire CDKN2A, CDK4, BAP1 Surveillance ciblée Prévention- Dépistage

89
Recommandations du groupe d’experts
Famille CDKN2A mutée, patient CDKN2A muté asymptomatique : - examen dermatologique tous les 6 mois en milieu hospitalier ; Idéalement, examen en vidéodermoscopie numérique à M0, M3 et M12 , puis annuelle et photographie corporelle totale annuelle. - surveillance sans limitation d’âge, à vie.

90
Le mélanome une maladie multifactorielle
• Multiples allèles de prédisposition • Faible pénétrance • Gènes de Pigmentation : MC1R, MATP, ASIP, TYR, TYRP1,… • Gènes de Réparation de l’ADN (ATM, POLH, ..) • Gènes Immunité (FAS, CASP8, …) • FDR cliniques: phototype I-II, cheveux clairs, Yeux clairs, nb élevé de nevus, taches de rousseur • Interaction avec l’exposition UV Familial Sporadique
 • Gènes Immunité (FAS, CASP8, …) • FDR cliniques: phototype I-II, cheveux clairs, Yeux clairs, nb élevé de nevus, taches de rousseur. • Interaction avec l’exposition UV. Familial. Sporadique.")
91
Melanocortin 1 receptor (MC1R)
Variants RHC= Red Hair Colour

92
Variants MC1R RHC et risque de mélanome
D84E - R142H - R160W - R151C - D294H MC1R genotype Cases Controls P-value OR [CI] 0/0 563 1052 reference 1/0 347 568 3.38 x 10-18 [ ] 1/1 52 77 [ ]

93
Analyses multivariées
Risk factor B P VALUE OR ICInf ICSup RHC 0,66 0,00012 1,94 1,38 2,72 NON RHC 0,32 0,042 1,37 1,01 1,87 sex -18,96 0,96 - ephelides -0,42 0,0098 0,48 0,90 sunburns < 15 0,60 0,0004 1,82 1,31 2,55 hair color -1,31 1,28E-05 0,27 0,15 skin type 0,17 1,19 0,87 1,62 Variants MC1R = FDR indépendant de mélanome +++

94
Variants MC1R et Mélanome Familial
Pénétrance Mutation CDKN2A + variant MC1R 80% Mutation CDKN2A + pas de variant MC1R 60% 40% CDKN2A non muté + variant MC1R 20 % CDKN2A non muté + pas de variant MC1R Age Age 10 20 30 40 50 60 70 80 90 35

95
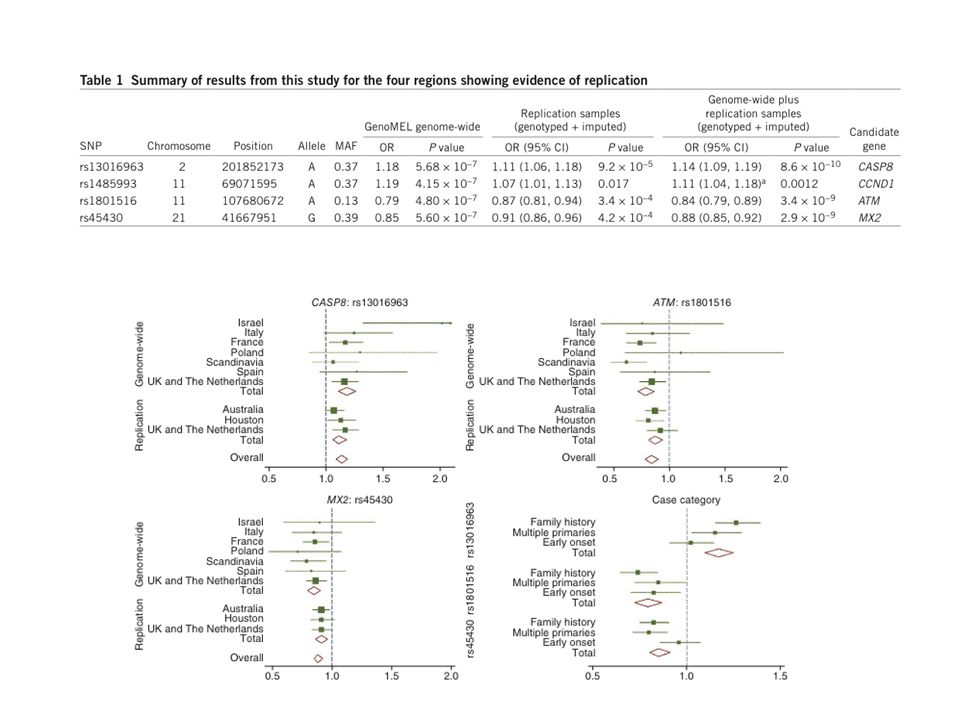
Genome-wide association study identifies three new melanoma susceptibility loci (Barett et al, Nat Genet, 2011) . GenoMEL Consortium 2,981 individuals, 6,426 control subjects Replication study : - 2 genome-wide studies (from Australia and Texas, USA) - UK and Netherlands Three loci replicated - ATM (rs , overall P = 3.4 × 10(-9)), - SNP in MX2 (rs45430, P = 2.9 × 10(-9)) - SNP adjacent to CASP8 (rs , P = 8.6 × 10(-10)) Un 4eme locus CCND1
 - UK and Netherlands. Three loci replicated. - ATM (rs , overall P = 3.4 × 10(-9)), - SNP in MX2 (rs45430, P = 2.9 × 10(-9)) - SNP adjacent to CASP8 (rs , P = 8.6 × 10(-10)) Un 4eme locus CCND1.")
97
Bilan des gènes impliqués dans la prédisposition multifactorielle au mélanome
MC1R SLC45A2 MITF TERT 20q TYR TYRP1 ASIP IRF4 ATM MX2 CASP8 9p21 1q21.3 rs PLAG2G6 OR de 0.35 à 6

98
Prédisposition au mélanome
monogénique CDKN2A, CDK4, BAP1 multifactorielle Variants fréquents Variants rares MM familial, multiple, + K pancréas MC1R SLC45A2 EDNRB, MITF…. TYR ASIP Conseil génétique Futurs biomarqueurs ? Surveillance sujets à risque

99
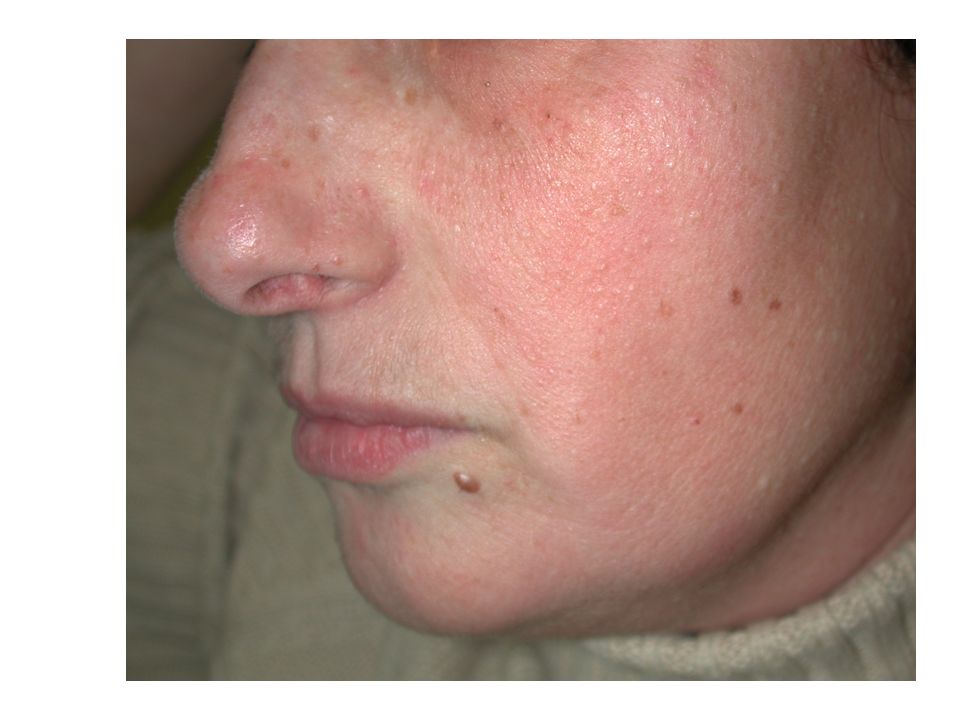
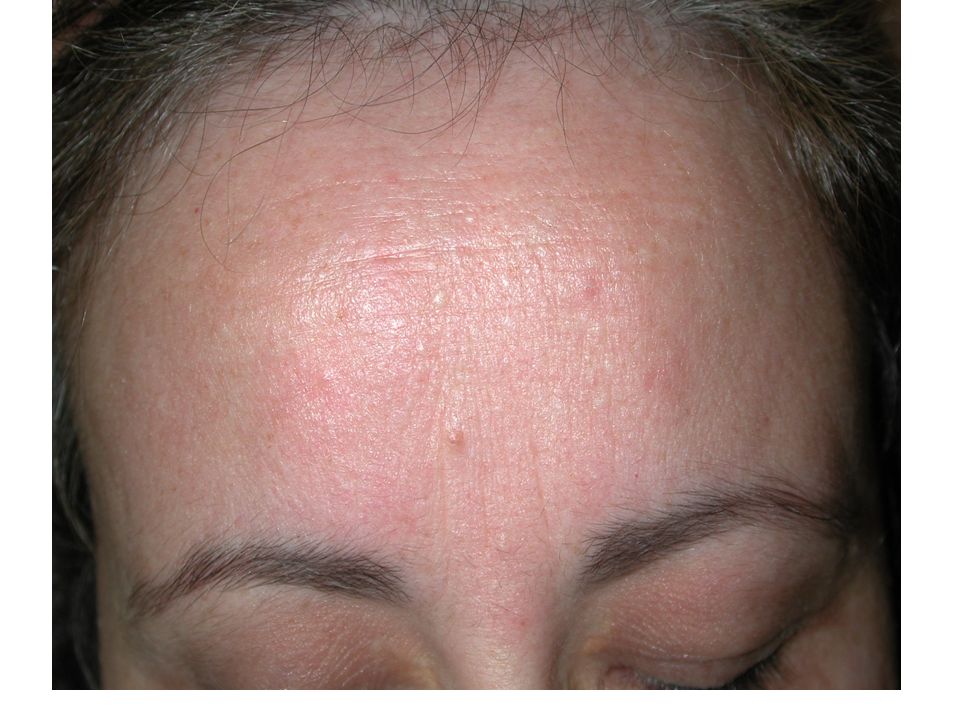
Syndrome de Birt Hogg Dubé
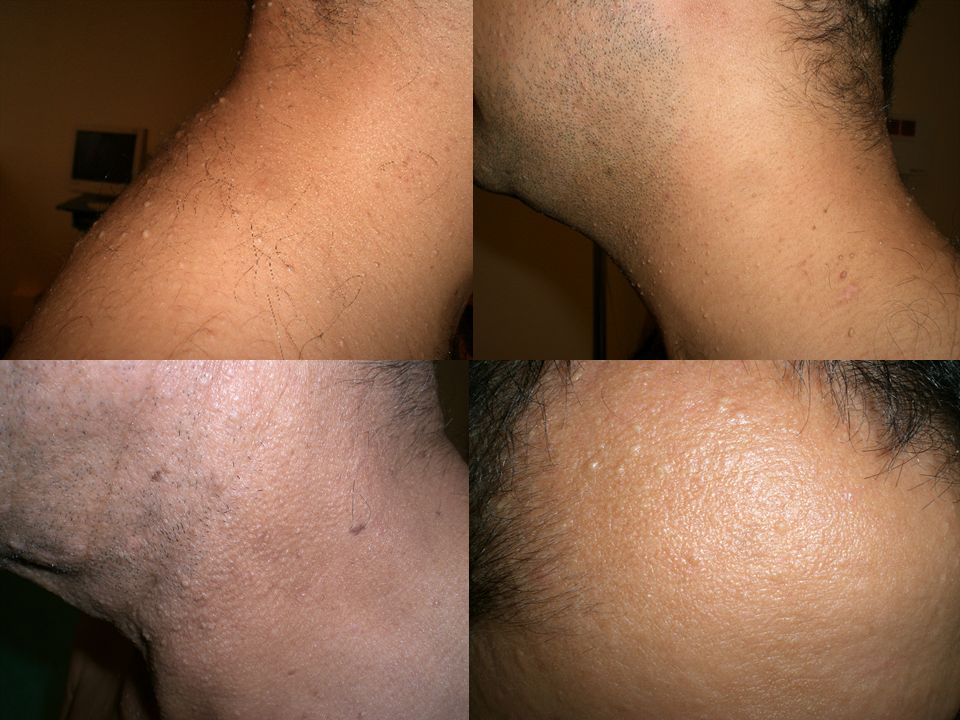
Valérie 99

102
Cordons de cellules basaloïdes
Follicule distordu Fibrose collagène Cordons de cellules basaloïdes 102

103
1) Quels examens demander ? Chez Valérie
Chez sa mère M-France, le reste de la famille 2) Comment organiser le conseil génétique ? - PAPULES BLANCHÂTRES
 Comment organiser le conseil génétique - PAPULES BLANCHÂTRES.")
104
Cas clinique: réponses
Chez Valérie (cas index) : + Test génétique après consentement éclairé en consultation d’oncogénétique + Scanner rénal, scanner thoracique, coloscopie En cas de mutation: Diagnostic pré-symptomatique chez ses apparentés, puis examens complémentaires en fonction du résultat Chez sa mère: TDM thoraco-abdominal et coloscopie
 : + Test génétique après consentement éclairé. en consultation d’oncogénétique. + Scanner rénal, scanner thoracique, coloscopie. En cas de mutation: Diagnostic pré-symptomatique chez ses apparentés, puis examens complémentaires en fonction du résultat. Chez sa mère: TDM thoraco-abdominal et coloscopie.")
105
Syndrome de Birt Hogg Dubé
• Transmission autosomale dominante • Fibrofolliculomes, des trichodiscomes, et des acrochordons • Tumeurs du rein: oncocytomes , tumeurs hybrides (chromophobes, papillaires, cellules claires, …) bilatérales ou multifocales • Pneumothorax et/ou emphysème bulleux • Cancers colo-rectaux. 105
 bilatérales ou multifocales. • Pneumothorax et/ou emphysème bulleux. • Cancers colo-rectaux")
106
examen dermatologique
Fibrofolliculomes parfois peu visibles examen dermatologique + histologie systématique

108
BHD Clinique (2) Tumeurs Bénignes - Goître multinodulaire,
- Adénome and oncocytome de la parotide - Trichoblastome - Mucinose, lipome, angiolipome - léiomyome cutané Tumeurs Malignes - Cancer sein, sarcome jambe - Cancer langue, cancer poumon, - Mélanome, CBC, carcinome épidermoïde, dermatofibrosarcome - Léiomyosarcome cutané

109
Emphysème bulleux

110
BHD physiopathologie FLN FLNC

111
BHD génétique Mutation germinale du gène de la folliculine retrouvée dans 50 % à 80% des cas chromosome 17p11.2 Point chaud dans l’exon 11 Délétions germinales dans 15-20% des cas

112
Bilan initial dans le BHD
Biopsie cutanée +++ Diagnostic moléculaire TDM abdominopelvien TDM thoracique initial Coloscopie 112

113
Conseil génétique Mutation identifié chez le cas index:
Diagnostic présymptomatique chez les apparentés +++ Si mutation: Scanner abdominopelvien initial puis echo ou TDM annuel Coloscopie tous les 2 ans Absence de mutation: pas de suivi particulier Mutation non identifiée chez le cas index: Surveillance par échographie et coloscopie de tous les apparentés 113

Présentations similaires







>")

 Biomarqueurs IHC (n = 412) Séquençage (n = 418) 200 patients évaluables pour les facteurs pronostiques cliniques et biologiques Comparaison.>")





>")
 Pr E. Tournier-Lasserve>")


 Pr E. Tournier-Lasserve>")



