Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Dr Francois Ghiringhelli Centre Georges Francois Leclerc Dijon
Thérapies ciblées - Biothérapies (thérapie génique, immunothérapie…) Etat actuel - Profil de tolérance - Perspectives Dr Francois Ghiringhelli Centre Georges Francois Leclerc Dijon
 Etat actuel - Profil de tolérance - Perspectives. Dr Francois Ghiringhelli. Centre Georges Francois Leclerc Dijon.")
5
1. Les thérapies ciblées

6
Plan 1/ La transduction du signal & cellules tumorales
2/ La transduction du signal & l’angiogénèse

7
Introduction:Transduction du Signal
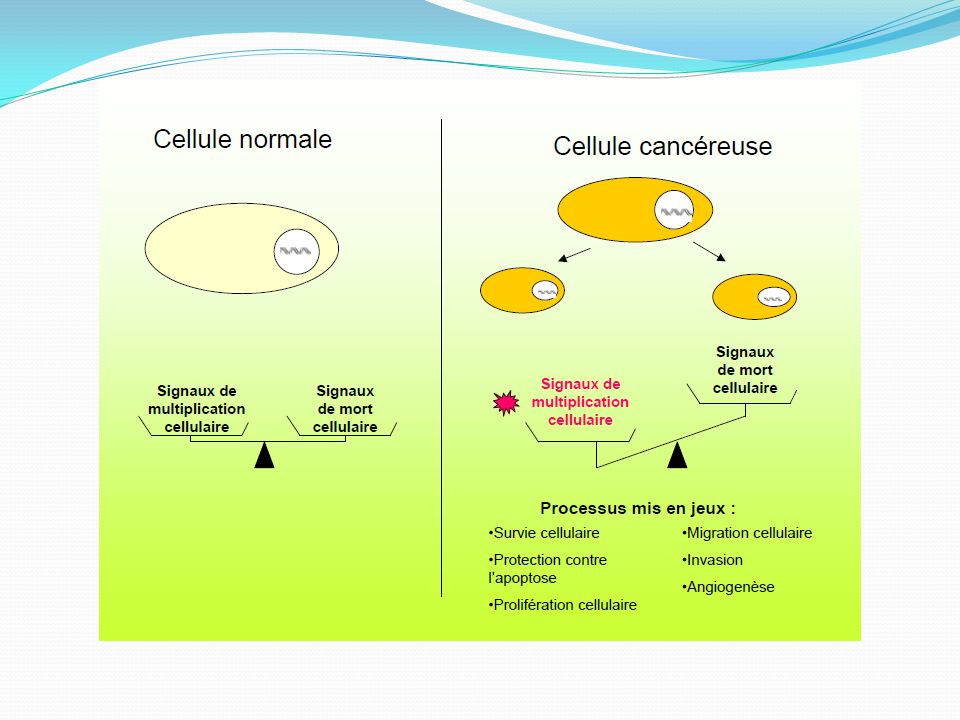
Le passage d’une cellule eucaryote de la phase quiescente (G0) à la phase de mitose (M), en réponse à des stimuli extérieurs est un processus multi-étape Il requiert la transduction de signaux divers et l’activation de protéines intracellulaires Ce processus finement régulé est altéré au sein des cellules néoplasiques, engendrant une prolifération, une migration et une différenciation cellulaire non contrôlée 3 étapes: Fixation sur un récepteur trans-membranaire d’un signal extra-cellulaire Activation de seconds messagers intra-cellulaires aboutissant à l’activation de facteurs de transcriptions qui vont pouvoir agir directement sur la transcription de gènes Cycle cellulaire
 à la phase de mitose (M), en réponse à des stimuli extérieurs est un processus multi-étape. Il requiert la transduction de signaux divers et l’activation de protéines intracellulaires. Ce processus finement régulé est altéré au sein des cellules néoplasiques, engendrant une prolifération, une migration et une différenciation cellulaire non contrôlée. 3 étapes: Fixation sur un récepteur trans-membranaire d’un signal extra-cellulaire. Activation de seconds messagers intra-cellulaires aboutissant à l’activation de facteurs de transcriptions qui vont pouvoir agir directement sur la transcription de gènes. Cycle cellulaire.")
8
Ras-GDP Ras-GTP Membrane Cytoplasme Noyau K HER-1 (EGFR) Homodimères
Hétérodimères PKC DAG PLCγ Gab-1 Endocytose IP3 HER PI3K Ras-GTP Ras-GDP Raf-1 Erk-2 Erk-1 Cytoplasme Shc Grb2 Sos K Dégradation lysosomale β1 α Importin α & β1 interaction Akt STATs C-Src Adhésion Migration FAK Facteurs de transcription Prolifération E2F1 STAT3 Prolifération Survie Survie Noyau Cycline D1

9
Récepteur Tyrosine Kinase R-TK
Domaine extracellulaire (liaison avec le ligand) Domaine transmembranaire Domaine cytoplasmique (Activité tyrosine kinase)
 Domaine. transmembranaire. Domaine cytoplasmique. (Activité tyrosine kinase)")
10
Les Récepteurs Trans-Membranaires,
des Cibles Thérapeutiques Il est actuellement décrit plusieurs familles de récepteurs tyrosine kinase (R-TK) qui se différencient par leur ligand et leur structure chimique mais qui possèdent des caractéristiques communes Famille de récepteur Classe I : ErbB / HER Classe II FGF Classe III HGF Classe IV Insuline Classe V Neurotrophins Classe VI PDGF Ligand EGF, TGF, AR, HB-EGF, SDGF, Heregulin, Betacellulin FGF 1 à 8 Insuline, IGF-1 IGF-2 NGF BDNF NT-3 à 5 PDGF-A et B VEGF CSF-1 SCF PIGF Récepteur HER-1 HER-2 HER-3 HER-4 FGFR-1 FGFR-2 FGFR-3 FGFR-4 MET IR IGFI-R TRKA TRKB TRKC PDGFR alpha et béta CSF-1R SCFR FLT KDR
 qui se différencient par leur ligand et leur structure chimique mais qui possèdent des caractéristiques communes. Famille de récepteur. Classe I : ErbB / HER. Classe II. FGF. Classe III. HGF. Classe IV Insuline. Classe V. Neurotrophins. Classe VI. PDGF. Ligand. EGF, TGF, AR, HB-EGF, SDGF, Heregulin, Betacellulin. FGF 1 à 8. Insuline, IGF-1. IGF-2. NGF. BDNF. NT-3 à 5. PDGF-A et B. VEGF. CSF-1. SCF. PIGF. Récepteur. HER-1. HER-2. HER-3. HER-4. FGFR-1. FGFR-2. FGFR-3. FGFR-4. MET. IR. IGFI-R. TRKA. TRKB. TRKC. PDGFR alpha et béta. CSF-1R. SCFR. FLT. KDR.")
11
HER-1 (EGFR) Homodimères HER-2 HER-3 HER-4 Hétérodimères Ras-GTP Ras-GDP Raf-1 Erk-2 Erk-1 Shc Grb2 Sos K 1/ Voie Ras/Raf/mitogen-activated protein kinase (MAPK) Erk = MAPK Facteurs de transcription Prolifération
 Erk = MAPK. Facteurs de. transcription. Prolifération.")
12
Ras-GDP Ras-GTP 2/ Voie Phosphatidylinositol 3-kinase/Akt K HER-1
(EGFR) Homodimères HER-2 HER-3 HER-4 Hétérodimères Gab-1 PI3K Ras-GTP Ras-GDP Raf-1 Erk-2 Erk-1 Shc Grb2 Sos K Akt 2/ Voie Phosphatidylinositol 3-kinase/Akt Facteurs de transcription Prolifération Survie
 Homodimères. HER-2. HER-3. HER-4. Hétérodimères. Gab-1. PI3K. Ras-GTP. Ras-GDP. Raf-1. Erk-2. Erk-1. Shc. Grb2. Sos. K. Akt. 2/ Voie Phosphatidylinositol 3-kinase/Akt. Facteurs de. transcription. Prolifération. Survie.")
13
R-TK (HER/IGF-R) Akt Acides Aminés PI 3-kinase PIP 3 PTEN PDK - T308
mTOR Cycle cellulaire - cyclin D1 - P27kip-1 P70 S6K 4E-BP1 eIF4G, eIF4B Recrutement des Ribosomes & Traduction des Protéines Raptor PI 3-kinase PIP 3 PTEN PDK Akt - T308 - S473 FKHR GSK3b BAD IKKa Survie cellulaire Apoptose

14
Ras-GDP Ras-GTP 3/ Voie Phospholipase Cγ K HER-1 (EGFR) Homodimères
Hétérodimères PLCγ IP3 DAG PKC Gab-1 PI3K Ras-GTP Ras-GDP Raf-1 Erk-2 Erk-1 Shc Grb2 Sos K Akt 3/ Voie Phospholipase Cγ Facteurs de transcription Prolifération Survie

15
Ras-GDP Ras-GTP 4/ Voie STAT
HER-1 (EGFR) Homodimères HER-2 HER-3 HER-4 Hétérodimères PKC DAG PLCγ Gab-1 IP3 PI3K Ras-GTP Ras-GDP Raf-1 Erk-2 Erk-1 Shc Grb2 Sos K Akt STATs 4/ Voie STAT Signal transducers and activator of transcription Facteurs de transcription Prolifération Survie
 Homodimères. HER-2. HER-3. HER-4. Hétérodimères. PKC. DAG. PLCγ. Gab-1. IP3. PI3K. Ras-GTP. Ras-GDP. Raf-1. Erk-2. Erk-1. Shc. Grb2. Sos. K. Akt. STATs. 4/ Voie STAT. Signal transducers and activator of transcription. Facteurs de. transcription. Prolifération. Survie.")
16
Ras-GDP Ras-GTP Src nine-gene family of nonreceptor TK
HER-1 (EGFR) Homodimères HER-2 HER-3 HER-4 Hétérodimères PKC DAG PLCγ Gab-1 IP3 PI3K Ras-GTP Ras-GDP Raf-1 Erk-2 Erk-1 Src nine-gene family of nonreceptor TK Shc Grb2 Sos K Akt STATs C-Src 5/ Voie Src kinase Adhesion Migration FAK Facteurs de transcription Prolifération Survie
 Homodimères. HER-2. HER-3. HER-4. Hétérodimères. PKC. DAG. PLCγ. Gab-1. IP3. PI3K. Ras-GTP. Ras-GDP. Raf-1. Erk-2. Erk-1. Src nine-gene family of. nonreceptor TK. Shc. Grb2. Sos. K. Akt. STATs. C-Src. 5/ Voie Src kinase. Adhesion. Migration. FAK. Facteurs de. transcription. Prolifération. Survie.")
17
Famille HER K Pas de ligand TGF alpha HB-EGF Ligand Binding Tyrosine
Epi TGF alpha HB-EGF Epi HB-GF NRG1 NRG2 NRG3 NRG4 BTC BTC NRG1 NRG2 EGF Pas de ligand AR Ligand Binding K Tyrosine Kinase EGFR HER1 C-erbB HER2 C-erbB2 HER3 HER4

18
(Liaison avec le ligand) (Activité tyrosine kinase)
R-TK: Dimérisation L’interaction avec un ligand induit une dimérisation du récepteur, élément critique pour l’initiation du signal intracellulaire. Les récepteurs dimérisés sont alors activés via à la fois une autophosphorylation ou une transphosphorylation transmoléculaire des résidus de type tyrosine kinase au niveau des domaines cytoplasmiques Domaine extracellulaire (Liaison avec le ligand) Domaine transmembrannaire Dimérisation Domaine cytoplasmique (Activité tyrosine kinase) 1 + 1 1 + 2
 Domaine. transmembrannaire. Dimérisation. Domaine cytoplasmique. (Activité tyrosine kinase)")
19
(Liaison avec le ligand) (Activité tyrosine kinase)
R-TK: Dimérisation HER-2 qui n’apparaît pas avoir de ligand est le partenaire favori de dimérisation des autres récepteurs HER. Les hétérodimères contenant HER -2 ont des caractéristiques particulières comme une dissociation avec le ligand et une endocytose plus lentes induisant un signal d’amont plus prolongé et plus puissant permettant une prolifération cellulaire plus importante. Domaine extracellulaire (Liaison avec le ligand) Domaine transmembrannaire Dimérisation Domaine cytoplasmique (Activité tyrosine kinase) 1 + 1 1 + 2
 Domaine. transmembrannaire. Dimérisation. Domaine cytoplasmique. (Activité tyrosine kinase)")
20
Activation de la transduction du signal
Les récepteurs TK, des cibles thérapeutiques Les mécanismes d’activation Mutation ex, EGFR; mutations au niveau de 4 exons TK, 18 – 19 – pas de liaison à un ligand, mais activation constitutive du domaine kinase Modifications génomiques avec hyperexpression du récepteur Augmentation de la production de EGF ou TGF-α par les celules tumorales: création d’une boucle autocrine entrainant une activation constitutive de ErbB-1 Activation de la transduction du signal Hyperexpression HER-2 ≈ 20% Cancers du sein Boucle autocrine

21
Les stratégies pour inhiber ErbB
1/ Des anticorps monoclonaux dirigés vers le domaine extracellulaire du récepteur peut être utilisé pour prévenir les liaisons avec le ligand. Cette approche peut ainsi moduler la signalisation, la dimérisation ou l’expression du récepteur au niveau de la surface cellulaire aussi bien que le cytotoxicité anticorps dépendante ou faisant intervenir le complément 2/ Des petites molécules se liant au domaine kinase peuvent inhiber la phosphorylation et l’activation de la voie de signalisation en amont 1 Ac monoclonal 2 Inhibiteur de kinase 3 Antagoniste 4/ Toxine ligand

22
Les stratégies pour inhiber ErbB
2 Inhibiteur de kinase 1 Ac monoclonal 4/ Toxine ligand 3 Antagoniste 3/ Des antagonistes des récepteurs peuvent être utilisés en bloquant de façon compétitive la liaison avec le ligand 4/ Des ligands ou des anticorps spécifiques des récepteurs peuvent être couplés à des toxines létales. Après liaison avec le récepteur, la toxine est internalisée et tue la cellule tumorale

23
Les récepteurs TK, des cibles thérapeutiques
Panitunumab . cetuximab HER-1 3 Antagoniste 1 Ac monoclonal 2 Inhibiteur de kinase 4/ Toxine ligand

24
Les récepteurs TK, des cibles thérapeutiques
3 Antagoniste 1 Ac monoclonal 2 Inhibiteur de kinase 4/ Toxine ligand trastuzumab pertuzumab HER-2

25
Les récepteurs TK, des cibles thérapeutiques
3 Antagoniste 1 Ac monoclonal 2 Inhibiteur de kinase 4/ Toxine ligand HER-1,2 Gefitinib Erlotinib Lapatinib …

26
Les récepteurs TK, des cibles thérapeutiques
3 Antagoniste 1 Ac monoclonal 2 Inhibiteur de kinase 4/ Toxine ligand TDM-1: Trastuzumab + Maytansine HER-2

27
La transduction du signal : les seconds messagers
Une fois le récepteur trans-membranaire activé, il va s’en suivre une cascade d’évènements. Une molécule adaptatrice va se fixer sur le site phosphorylé du récepteur via un domaine d’homologie SH2. Cette molécule Shc activée va activer à son tour une protéine G intracytoplasmique. Cette protéine G va recruter une protéine « d’échange de guanine nucléotide » la protéine SOS 1 Grb2 Sos Shc PI3K Akt Ras Raf MEK1/2 MAPK BAD Survie Prolifération PTEN mTOR Progression du cycle cellulaire FKHR GSK3 p27 Cyclin D1, E

28
X La voie Ras/Raf/MEK/MAPK-ERK Molécules agissant sur Ras Grb2 Sos Shc
PI3K Akt Ras Raf MEK1/2 MAPK BAD Survie Prolifération PTEN mTOR Progression du cycle cellulaire FKHR GSK3 p27 Cyclin D1, E X

29
X La voie Ras/Raf/MEK/MAPK-ERK Inhibiteurs de Raf Kinase et de MEK
Grb2 Sos Shc PI3K Akt Ras Raf MEK1/2 MAPK BAD Survie Prolifération PTEN mTOR Progression du cycle cellulaire FKHR GSK3 p27 Cyclin D1, E X ISIS 5132 BAY PLX 4720 X CI-1040

30
X + La voie PI3K/PTEN/AKT/mTor
Inhibition de la PI 3Kinase et restauration de PTEN Grb2 Sos Shc PI3K Akt Ras Raf MEK1/2 MAPK BAD Survie Prolifération PTEN mTOR Progression du cycle cellulaire FKHR GSK3 p27 Cyclin D1, E X +

31
La voie PI3K/PTEN/AKT/mTor
Inhibiteurs de mTor Grb2 Sos Shc PI3K Akt Ras Raf MEK1/2 MAPK BAD Survie Prolifération PTEN mTOR Progression du cycle cellulaire FKHR GSK3 p27 Cyclin D1, E X Temsirolimus Everolimus

32
4/ Les inhibiteurs du cycle cellulaire
Grb2 Sos Shc PI3K Akt Ras Raf MEK1/2 MAPK BAD Survie Prolifération PTEN mTOR Progression du cycle cellulaire FKHR GSK3 p27 Cyclin D1, E PTEN X Flavopiridol UCN-01 E7070

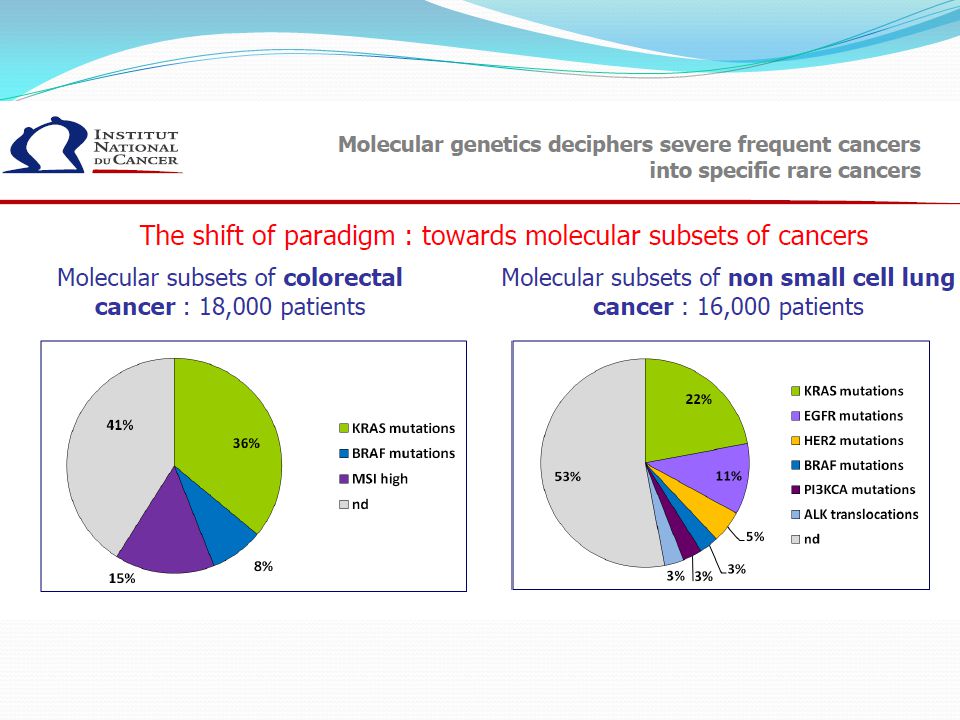
33
mutation exon 9 or secondary mutations
Tumor Type Driven Genomic Pathway Biomarker Rationale Therapeutic Response Advanced disease Biomarker of Resistance Breast HER2 (15-20%) Trastuzumab* Trastuzumab + CT1 ? Ongoing Lapatinib* Pertuzumab Neratinib T-DM1 GIST c-Kit (CD117) mutation exon 11 Imatinib* Yes1 c-Kit mutation exon 9 or secondary mutations exons 13-14 mutation exon 17 PDGFRA D842V mutation Imatinib 800 mg or sunitinib* Nilotinib CP-868,596 Melanoma BRAF V600E Mutation (50%) PLX4720 NRas mutation MAP3K mutation COT upregulation ERK inhib. *AMM publication (PubMed) Italique (AACR 2011)
 Trastuzumab* Trastuzumab + CT1. Ongoing. Lapatinib* Pertuzumab. Neratinib. T-DM1. GIST. c-Kit (CD117) mutation exon 11. Imatinib* Yes1. c-Kit. mutation exon 9 or secondary mutations. exons mutation exon 17. PDGFRA D842V mutation. Imatinib 800 mg. or. sunitinib* Nilotinib. CP-868,596. Melanoma. BRAF V600E Mutation (50%) PLX4720. NRas mutation. MAP3K mutation. COT upregulation. ERK inhib. *AMM 1publication (PubMed) Italique (AACR 2011)")
34
Advanced Basal Cell Carcinoma BCC
Tumor Type Driven Genomic Pathway Biomarker Rationale Therapeutic Response Advanced disease Biomarker of Resistance Colon Kras Wild-Type Cetuximab* Yes1 BRAF mutation Lung Adenocarcinoma Mutations activ. EGFR (15%) exons 19 & 21 EML4/ALK fusion (5%) Gefitinib* Crizotinib EGFR mutation T790 ALK mutations L1196M & C11564 BIBW-2992 PF WZ 4002 X-396 HSP90 inh. sqamous cell DDR2 mutation FGFR1 amplification (20%) Dasatinib (Src inh.) FGFR inbh. Advanced Basal Cell Carcinoma BCC PITCH/SMO mutation GDC-0449* Hedgehog inhib. *AMM publication (PubMed) Italique (AACR 2011)
 exons 19 & 21. EML4/ALK. fusion (5%) Gefitinib* Crizotinib. EGFR mutation T790. ALK mutations. L1196M & C BIBW PF WZ X-396. HSP90 inh. sqamous cell. DDR2 mutation. FGFR1 amplification (20%) Dasatinib. (Src inh.) FGFR inbh. Advanced Basal Cell Carcinoma BCC. PITCH/SMO. mutation. GDC-0449* Hedgehog inhib. *AMM 1publication (PubMed) Italique (AACR 2011)")
37
En pratique: Cible inconnue par de biomarker (ex sunitinib, sorafenib)
2. Cible connue et hyperexprimée (HER2) 3. Cible mutante et sensible au médicament (Braf, EGFR) 4. Cible connue mutation en aval (Kras)
 3. Cible mutante et sensible au médicament (Braf, EGFR) 4. Cible connue mutation en aval (Kras)")
38
L ’angiogénèse tumorale: un processus complexe (1)
Incluant plusieurs étapes successives 1/ Production de facteurs de croissance angiogénique 2/ Activation des cellules endothéliales par ces facteurs 3/ Hyperperméabilité des vaisseaux sanguins 4/ Dégradation des membranes basales et invasion par les cellules endothéliales 5/ Prolifération des cellules endothéliales 6/ Stabilisation des nouveaux vaisseaux sanguins via la migration des péricytes 7/ Croissance tumorale et métastases

39
L ’angiogénèse tumorale: un processus complexe (2)
Chaque étape fournit une opportunité pour une intervention thérapeutique

40
3 1 2 Facteurs impliqués dans la promotion de l'Angiogénèse Tumorale
Cellules Stromales Stress métabolique Stress mécanique Inflammation Altération génique Cellules Tumorales 2 PDGF bFGF Cellules Endothéliales 1 TGFa Globules Rouges Péricytes Vaisseaux Sanguins

41
MODULATEURS DE L’EXPRESSION DE VEGF
TGF Il1β, Il6 PGE2 COX2 EGF FGF IGF PDGF P53 VHL VEGF Angiogenèse + Ras Raf Stress oxydatif Hypoxie HIF-1 MODULATEURS DE L’EXPRESSION DE VEGF - Heat shock Inducing Factor

42
Neutralisation des facteurs angiogèniques
Inhibition VEGF et VEGF-R

43
Transduction du signal activée par VEGF
VEGF Trap bevacizumab Cellules Stromales Cellules Tumorales Prolifération Différentiation Dégradation enzymatique Survie Migration Facteurs de transcription Protéases Protéines de structure Métabolisme enzymatique Angiogénèse Cellules Endothéliales

44
AVASTIN™ (AC RECOMBINANT HUMANISÉ ANTI VEGF)
93% humain, 7% murin Reconnait tous les isoformes du VEGF Kd = 8 x M Demi-vie jours

45
Inhibiteur de l’activité TK intra cellulaire : VEGFR

46
Inhibition spécifique de l'activité de VEGFR's
Anti-VEGF Cellules Stromales RTK VEGFR Inhibiteurs SU5416 Vatanalib AGO13736 AGO28262 GW786034 (VEGR1-2-3) IMC-1121B VEGFR1 IMC-18F1 VEGFR2 IMC-3C5 VEGFR3 Ac Anti-VEGFR Cellules Tumorales Inhibiteurs de la transduction du signal Prolifération Différentiation Dégradation enzymatique Survie Migration Facteurs de transcription Protéases Protéines de structure Métabolisme enzymatique Angiogénèse Cellules Endothéliales
 IMC-1121B VEGFR1. IMC-18F1 VEGFR2. IMC-3C5 VEGFR3. Ac. Anti-VEGFR. Cellules. Tumorales. Inhibiteurs de la transduction du signal. Prolifération. Différentiation. Dégradation. enzymatique. Survie. Migration. Facteurs de transcription. Protéases. Protéines de structure. Métabolisme enzymatique. Angiogénèse. Cellules. Endothéliales.")
47
Inhibition de plusieurs facteurs de croissance RTK
C-kit, autres kinases…. Cellules Tumorales Exemple; Sunitinib …. RTK Inhibiteurs à large spectre Péricytes Cellules Endothéliales FGFR Inh VEGFR Inh PDGFR Inh

48
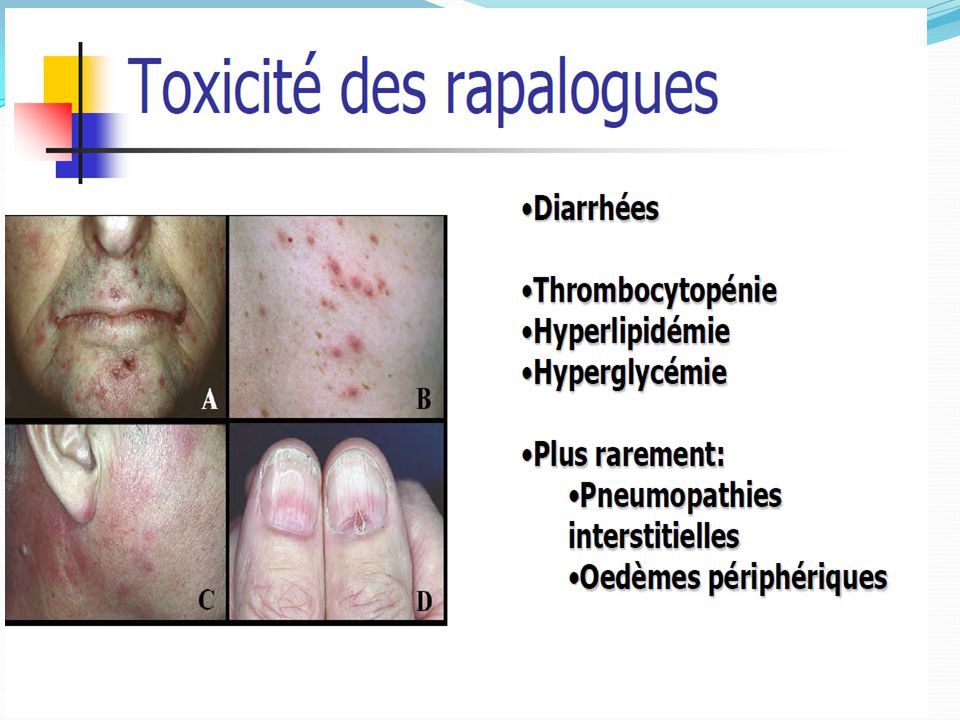
2. Profil de tolérance

62
3. La thérapie génique dans le glioblastome

63
Introduction BUT: insérer un gène dans la cellule malade dans le but de reverser le phénotype

64
Distribution of gene-transfer clinical protocols approved by or submitted to regulatory authorities in North America and Europe Disease Number Vector Number of of protocols protocols Cancer Retrov Infectious diseases Ad Monogenic diseases AAV Cardivascular diseases Poxviruses Rheumatoid arthritis HSV Cubital tunnel syndrome Naked DNA Total Lipids In vivo transfer Gene gun Ex vivo transfer Electroporation Naked RNA Total

65
La thérapie génique reste une technique expérimentale
Le problème majeur repose sur la capacité à transférer le gène dans la cellule cible Les resultats sont actuellement modestes.

66
SPECIFICITE DU CNS La barrière hémato-encéphalique
Absence de promoteurs spécifiqu e des cellules gliales Necessité d’infecter toutes les cellules

67
Méthode d’infection Le plus performant est l’injection du traitement via une injection stéréotaxique. Adénovirus HSV (utile pour la technique du gène suicide) Retrovirus Lentivirus (HIV Like) ADN nu Liposome
 Retrovirus. Lentivirus (HIV Like) ADN nu. Liposome.")
68
3 types de mechanismes Correction d’une anomalie génique: ex induction de p53

69
Infection avec un virus contenant un gene suicide

70
Utilisation d’un virus Oncolytique

71
4. Immunothérapie et glioblastome
Les différents types d’immunothérapie: Injection d’adjuvant un situ (Cpg) Vaccin contre un Ag de tumeur (EGFRvIII) Transfert de lc T (abandonné)
 Vaccin contre un Ag de tumeur (EGFRvIII) Transfert de lc T (abandonné)")
74
5. Thérapies ciblées en neurooncologie
V Lorgis

76
Communication entre vaisseaux et le SNC
Lu P, Werb Z : Science 2008;

77
Hypothèse du Switch Angiogenique
Brem S. Clin Neurosurg 1975

78
Le glioblastome est une tumeur vasculaire
Brem S, Cotran R, Folkman J, JNCI, 1972

79
Antiangiogénique d’usage clinique
Bevacizumab (Avastin™) Sunitinib (Sutent™) Sorafenib (Nexavar™) Cediranib (Recentin™ - AZD- 2171) (inhib VEGFR) Cilengitide (Inhibe integrin alphaV béta 3)
 Sunitinib (Sutent™) Sorafenib (Nexavar™) Cediranib (Recentin™ - AZD- 2171) (inhib VEGFR) Cilengitide (Inhibe integrin alphaV béta 3)")
80
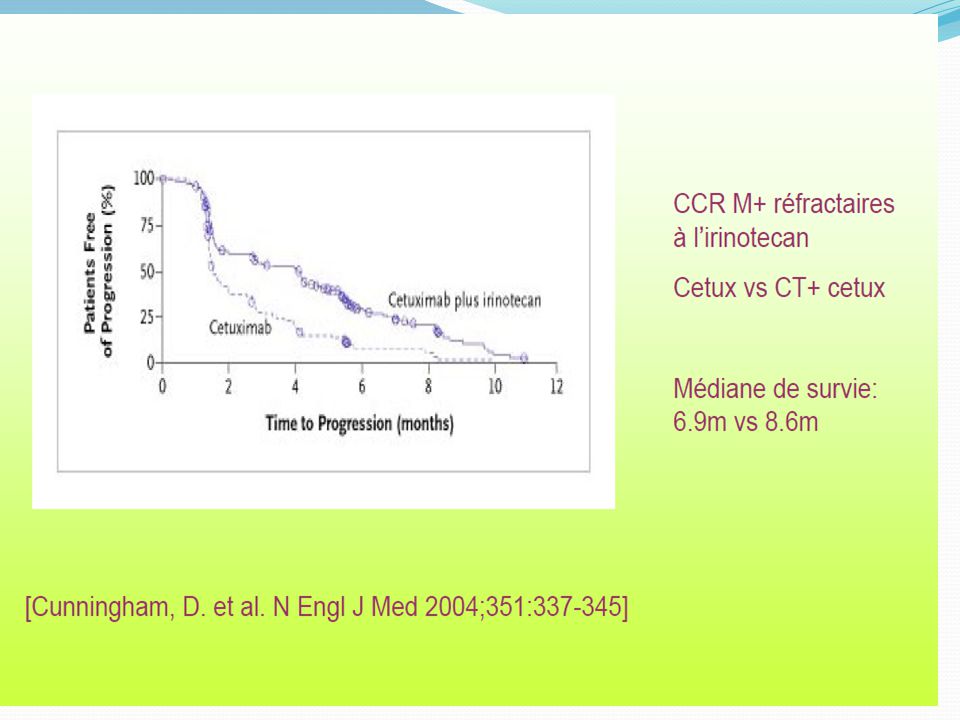
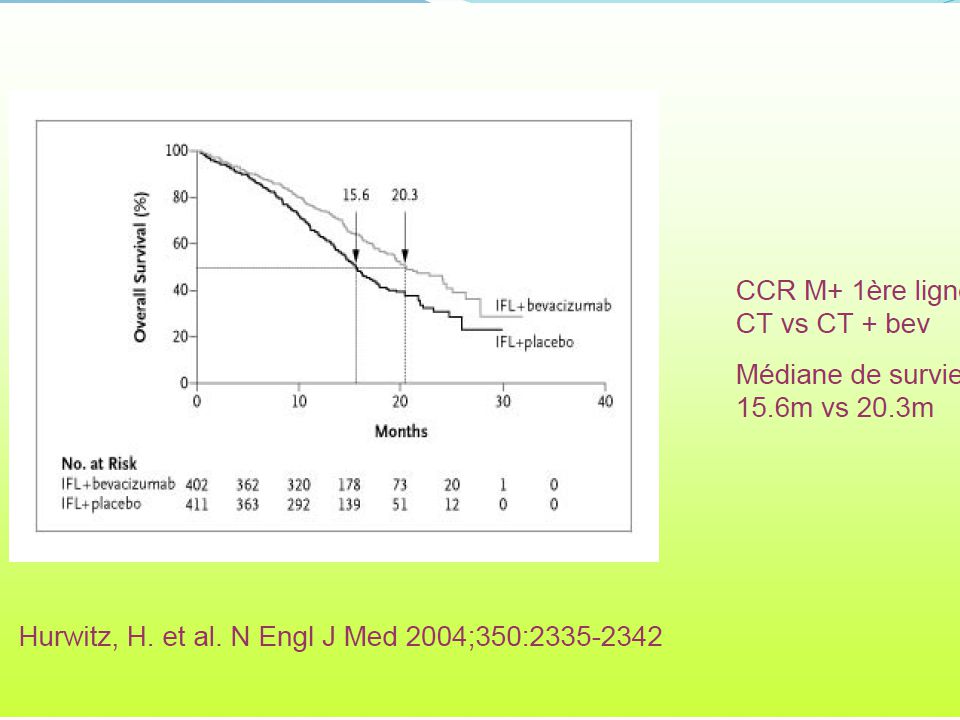
Bevacizumab- Efficacy in Clinical Trials – Metastatic Colorectal Cancer
From Ferrara N, Nat Rev Drug Discovery, 2004; Hurwitz et al, NEJM, 2004

81
Batchelor T, Brem S, Sorensen G, ANGIOGENESIS FOUNDATION, 2008

82
Mécanisme d’action Folkman J, Nature Drug Discovery 6:274, 2007

83
Bevacizumab + Irinotecan

84
AVANT ET 2 MOIS APRES Courtesy Dr. Sajeel Chowdhary, Moffitt Cancer Center

85
Taux de réponse Outcome Drug Bev AZD2171 NABTC Targeted Enzastaurin
CCNU Median PFS, wks 23 17 6-8 6 Median TTS 39 32 26 25 30.5 6-mo PFS, % 43 9 15 19 Response rate, % 28 56 NA 3 4 Bev, bevacizumab; CCNU, lomustine; GBM, glioblastoma multiforme; NA, not applicable; NABTC, North America Brain Tumor Coalition; PFS, progression-free survival; TTS, time to survival; VEGF, vascular endothelial growth factor. With anti‑VEGF therapy, these outcomes have changed, and the 2 particular therapies that have been well studied to date include bevacizumab and AZD2171, which is also known as cediranib. The data in the red box show improvements in progression‑free survival, time to survival, 6‑month progression‑free survival, and response rates. Therefore, these outcomes are really dramatically improved with the use of anti‑VEGF therapy. Wagner SA, et al. ASCO Abstract 2021. Batchelor T, et al. AACR Abstract LB-247. Fine HA, et al. ASCO Abstract 2005.

86
Bevacizumab ± Irinotecan : Phase II
Outcome Bev Alone (n = 85) Bev + Irinotecan (n = 82) Median OS, mos (95% CI) 9.2 ( ) 8.7 ( ) PFS at 6 mos, % (97.5% CI) 42.6 ( ) 50.3 ( ) Median PFS, mos (95% CI) 4.2 ( ) 5.6 ( ) ORR, % (97.5% CI) 28.2 ( ) 37.8 ( ) CR 1.2 2.4 PR 27.1 35.4 Median duration of response, mos (95% CI) 5.6 ( ) 4.3 (4.2-NR) Bev, bevacizumab; CI, confidence interval; CR, complete response; GBM, glioblastoma multiforme; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; PR, partial response. This slide displays findings from a phase II study that evaluated bevacizumab vs bevacizumab plus irinotecan. Results showed that the median survival was essentially the same between the treatment arms, and 6‑month progression‑free survival favored the bevacizumab plus irinotecan arm at 50% vs 43% for the bevacizumab alone arm. The median progression‑free survival also favored the irinotecan arm. The response rate slightly favored the irinotecan arm, and the duration of response slightly favored the bevacizumab alone arm. Friedman HS, et al. J Clin Oncol. 2009;27:
 Bev + Irinotecan (n = 82) Median OS, mos (95% CI) 9.2 ( ) 8.7 ( ) PFS at 6 mos, % (97.5% CI) 42.6 ( ) 50.3 ( ) Median PFS, mos (95% CI) 4.2 ( ) 5.6 ( ) ORR, % (97.5% CI) 28.2 ( ) 37.8 ( ) CR PR Median duration of response, mos (95% CI) 5.6 ( ) 4.3 (4.2-NR) Bev, bevacizumab; CI, confidence interval; CR, complete response; GBM, glioblastoma multiforme; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; PR, partial response. This slide displays findings from a phase II study that evaluated bevacizumab vs bevacizumab plus irinotecan. Results showed that the median survival was essentially the same between the treatment arms, and 6‑month progression‑free survival favored the bevacizumab plus irinotecan arm at 50% vs 43% for the bevacizumab alone arm. The median progression‑free survival also favored the irinotecan arm. The response rate slightly favored the irinotecan arm, and the duration of response slightly favored the bevacizumab alone arm. Friedman HS, et al. J Clin Oncol. 2009;27:")
87
Mécanisme de resistance

88
Echec du Beva 40% de non répondeurs
Pour les répondeures la récurrence donne lieu à un changement de phénotype: la gliomatose diffuse

89
40 ans homme GBM.6.2008 IRM preop T-1 Gd.

90
4 Mois post-op. RTX + temo. KPS-90

91
MAIS apparition d’un contraste. KPS-90

92
6 Mois postop – Confirmation de la récurrence

93
Image a 2 mois de CPT-11+ bevacizumab.

94
A 4 mois RC en T1 gado mais anomalie en FLAIR

95
A 6 mois toujours RC en gado mais augmentation du FLAIR

96
A 10 mois de Beva: ,progression clinique et radiologique en T2 mais pas de T1 gado

97
Une possibilité: l’apparition d’autres proangiogénique
Folkman J, Nature Drug Discovery 6:274, 2007

98
VEGF : facteur limitant l’invasion des gliomes
Du R et al, HIF1alpha induces recruitment of BMDC to regulate tumor angiogenesis and invasion. Cancer Cell 13: , 2008.

99
En conclusion: l’inibitiond e l’angiogénèse favorise l’invasion
Du R et al: Cancer Cell 13: , 2008.

100
The Clinical and Biological Imperative- Specific, Immediate, and Long-Term Objective
“ It will, therefore, be instrumental to identify pathways that simultaneously block perivascular invasion and angiogenesis to improve current antiangiogenic therapy in GBM and potentially other tumors”. Du R et al: Cancer Cell 13: , 2008.

Présentations similaires































 DANS LA PRISE EN CHARGE D’UNE TUMEUR DU SEIN Dr Olivier RENAUD Service d’anatomie-pathologique.>")

