Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Glomérulopathies

2
Classification des GN

3
Classification des GN

4
GN primitives non prolifératives

5
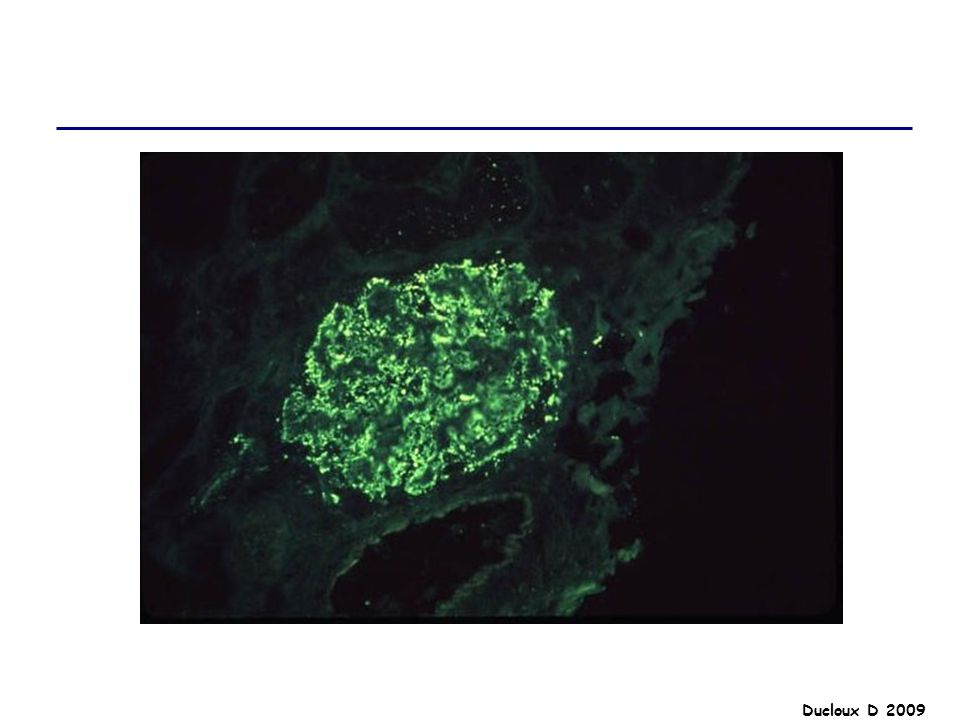
LGM Cause la plus fréquente de SN chez l’enfant entre 3 et 6 ans
80% des SN de l’enfant / 20% chez l’adulte Terrain atopique Vaccination, infection virale Maladie de Hodgkin / AINS A2 B8 DR3

6
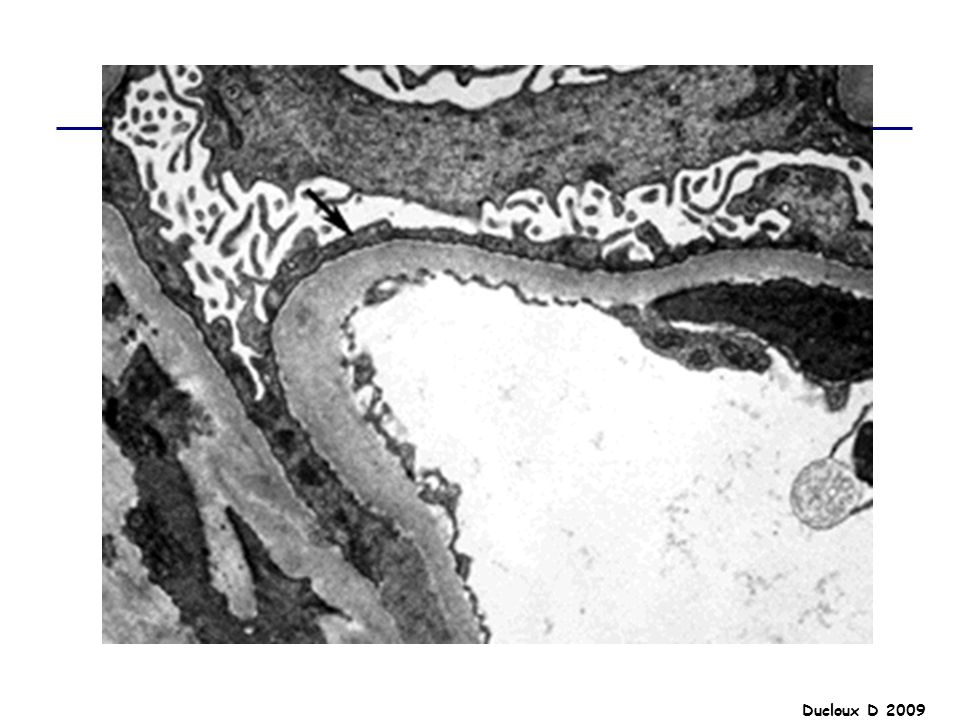
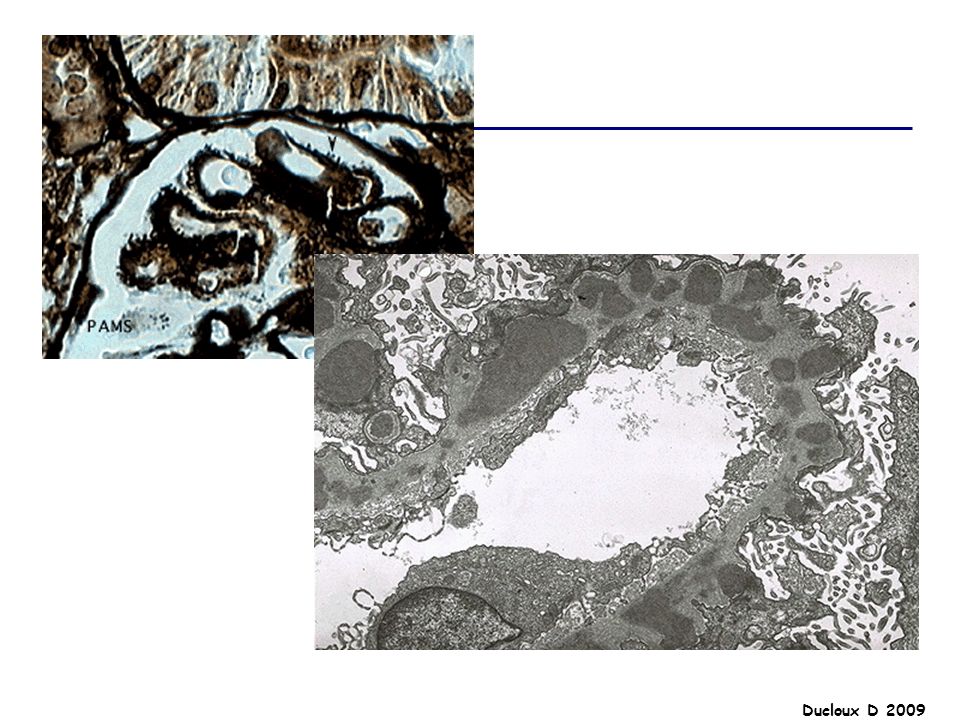
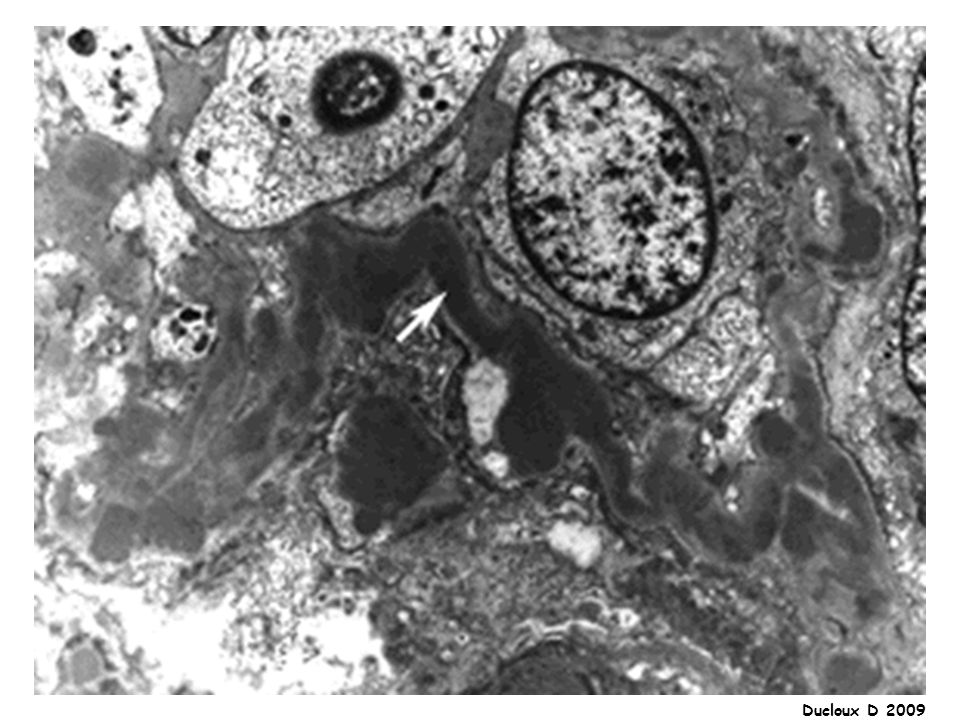
LGM Début brutal SN pur PBR : Microscopie optique : normale
IF négative Microscopie électronique : lésions épithéliales diffuses avec effacement ou fusion des pédicelles Indiquée chez l’adulte / Pas d’indication chez l’enfant de moins de 10 ans

8
LGM Physiopathologie Immunologique Activation lymphocytaire Th2
Lymphokine augmentant la perméabilité glomérulaire Efficacité des stéroïdes et des immunosuppresseurs

10
LGM Stéroïdes Cortico-résistance Cortico-dépendance Rechutes
1 à 2 m/kg/j pendant 8 à 10 semaines Décroissance progressive : Diminution de moitié de la posologie pour 4 semaines Diminution pour arrêt (~ 9 mois de traitement) Taux de rémission ~ 85% Récidive : 70% chez l’adulte Cortico-résistance Cortico-dépendance Rechutes Traitement non spécifique Traitement du SN Traitement accompagnant la corticothérapie
 Taux de rémission ~ 85% Récidive : 70% chez l’adulte. Cortico-résistance. Cortico-dépendance. Rechutes. Traitement non spécifique. Traitement du SN. Traitement accompagnant. la corticothérapie.")
11
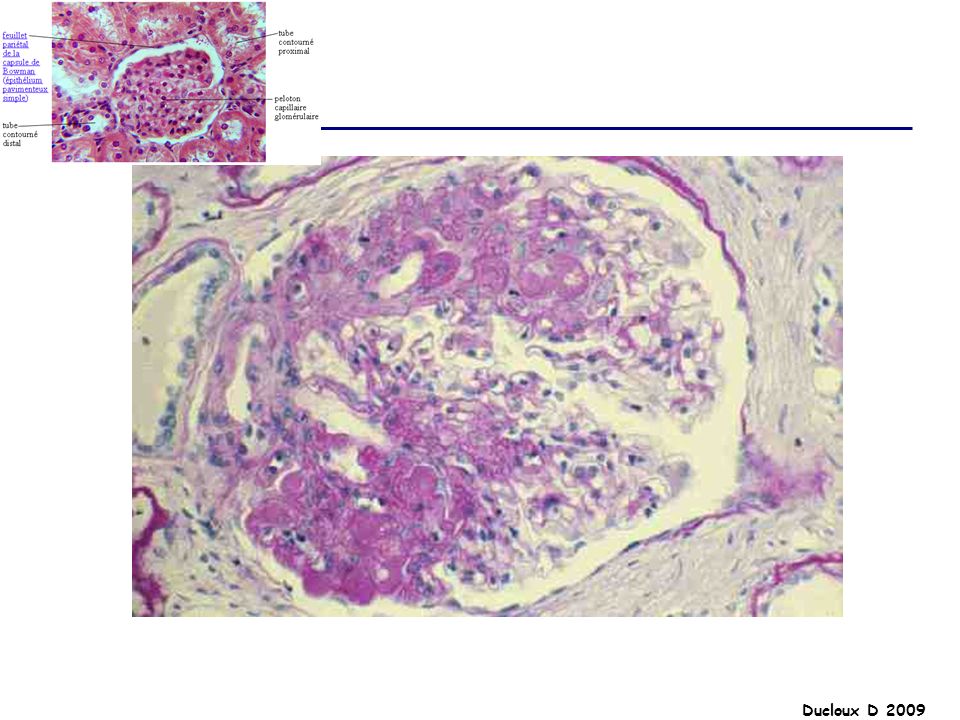
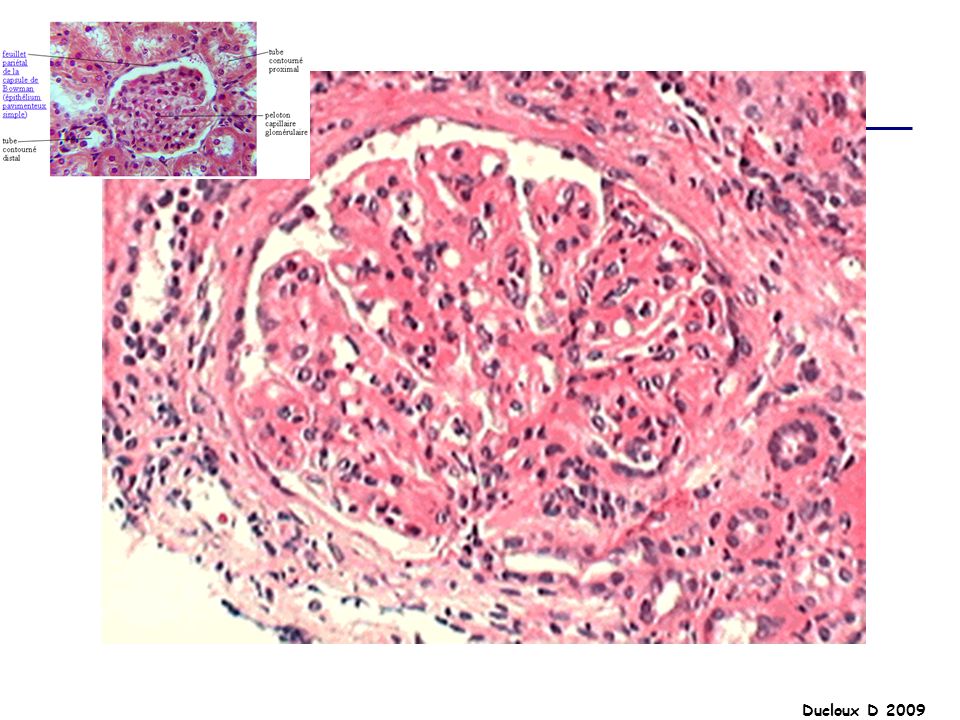
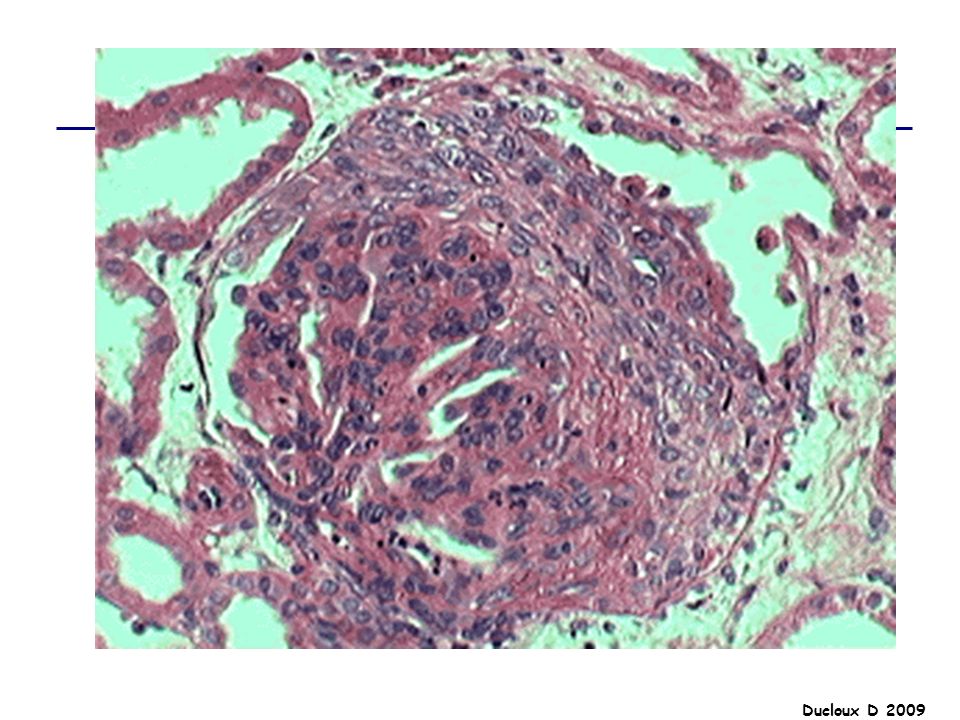
HSF Ensemble d’affections glomérulaires diverses partageant une lésion histologique commune En microscopie optique : collapsus glomérulaire adhésion du floculus à la membrane de Bowman condensation de matériel hyalin amorphe +/- hypercellularité mésangiale modérée et occlusion partielle de la lumière capillaire par les dépôts hyalins LESIONS : SEGMENTAIRES FOCALES

14
HSF La HSF représente environ 5 à 15% des causes de SN de l’adulte et moins de 10% des SN de l’enfant Elle est plus fréquente chez le sujet de race noire, et chez l’homme SN dans 70% des cas SN impur HTA 50% Hématurie microscopique 50% Insuffisance rénale 20 à 30%

15
HSF Idiopathique Secondaire Reflux vésico-urétéral HIV
Réduction néphronique

16
HSF Pronostic IR d’emblée SN résistant à la corticothérapie
Fibrose interstitielle > 20% Lésions cellulaires

17
HSF Stéroïdes Cortico-résistance 1 mg/kg/j pendant 12 à 16 semaines
Décroissance très progressive Cortico-résistance

18
GEM Maladie de l’adulte
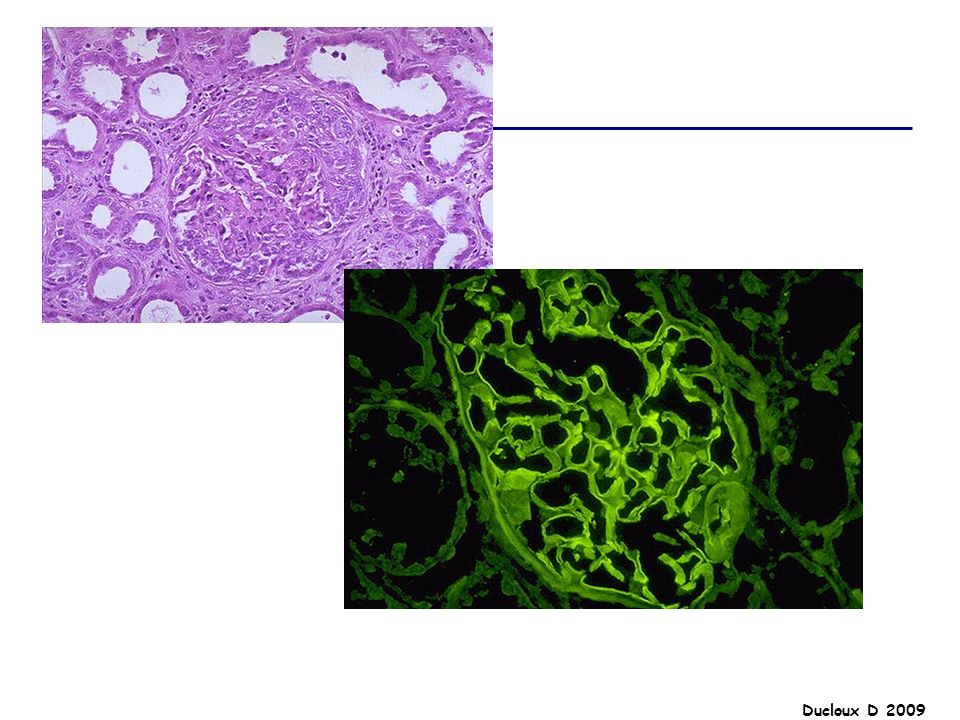
Le syndrome néphrotique est le mode de révélation le plus fréquent de la GEM (80%) L'hypertension est présente dans environ 30 % des cas, l'hématurie microscopique dans 50 % des cas La GEM est définie histologiquement par la présence des dépôts d'immunoglobulines G, situés sur le versant externe de la membrane basale glomérulaire en position sous-épithéliale
 L hypertension est présente dans environ 30 % des cas, l hématurie microscopique dans 50 % des cas. La GEM est définie histologiquement par la présence des dépôts d immunoglobulines G, situés sur le versant externe de la membrane basale glomérulaire en position sous-épithéliale.")
19
Spike

24
GEM La GEM est fréquemment secondaire (25%) et un bilan étiologique doit être conduit à la recherche des principales étiologies
 et un bilan étiologique doit être conduit à la recherche des principales étiologies.")
25
GEM Dans les formes primitives l’évolution est très variable
Rémission spontanée de la protéinurie : 5 à 20 % des patients Une rémission partielle définie par une protéinurie inférieure à 2 g/jour survient chez 25 à 40 % des patients. L'incidence de l'insuffisance rénale terminale est d'environ 15 % à 5 ans, 35 % à 10 ans et 40 % à 15 ans Facteurs de mauvais pronostic sexe masculin, âge supérieur à 50 ans, l’intensité et la durée d’évolution du SN, et l’existence d’une fibrose interstitielle sur la biopsie.

26
GEM Traitement Abstention Alkylants + stéroïdes Ciclosporine Autres

27
GN primitives prolifératives

28
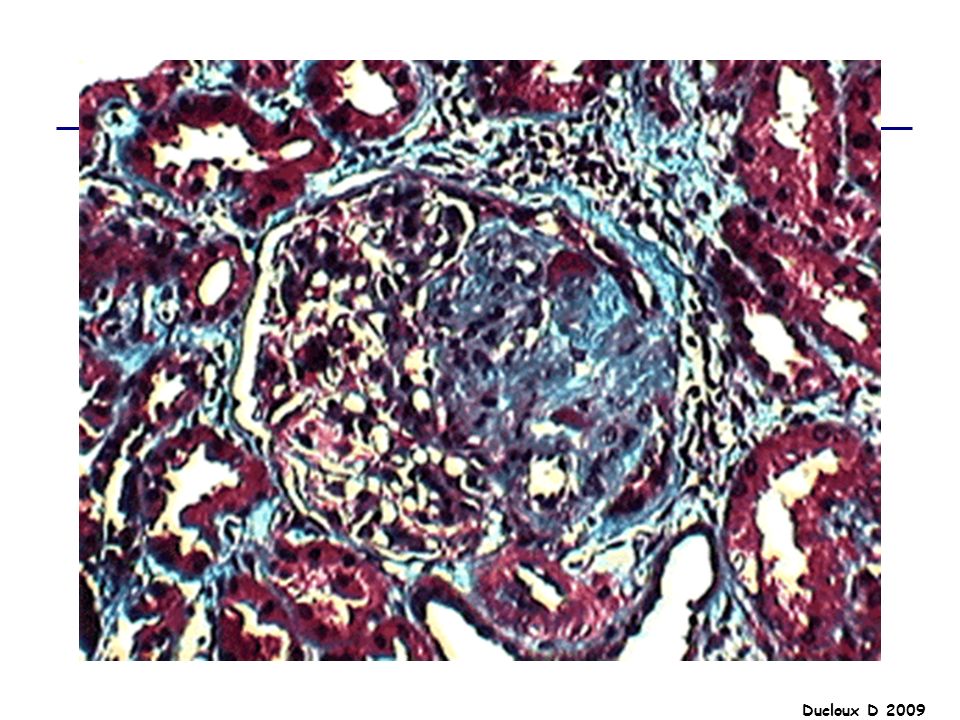
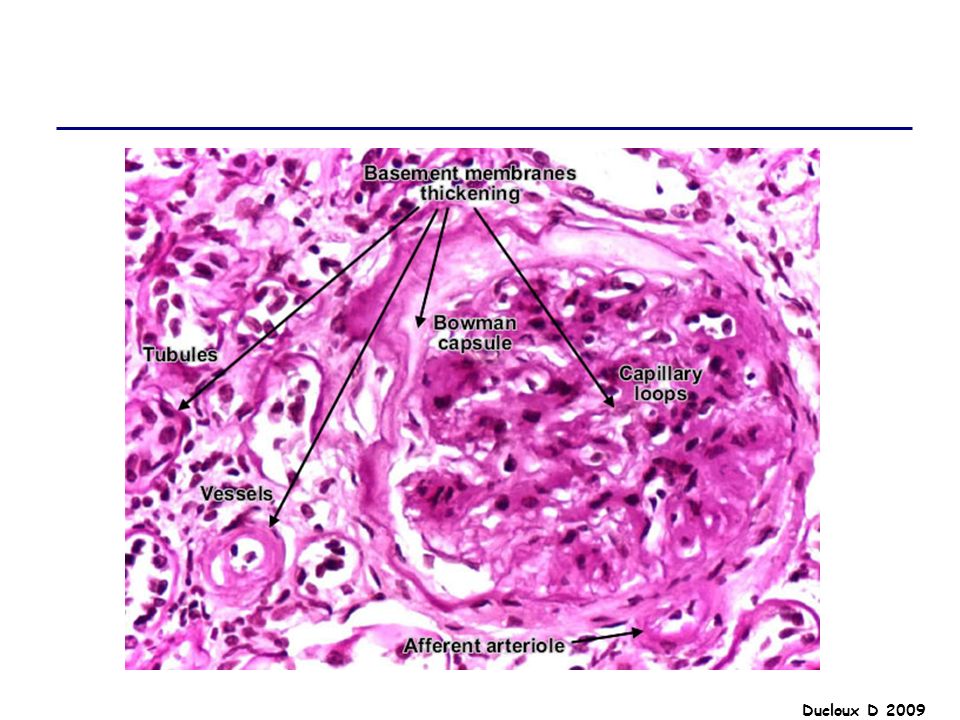
GNMP La GNMP est caractérisée par des lésions histologiques qui recouvrent des affections diverses. Lésion histologique commune microscopie électronique : épaississement de la MBG, en rapport avec des dépôts d’immuns complexes et avec l’interposition de cellules mésangiales entre la MBG et les cellules endothéliales. Il existe par ailleurs une hypercellularité, liée à la prolifération des cellules mésangiales et à l’afflux de monocytes, qui aboutit à un aspect lobulé du floculus.

29
Afflux monocytes Interposition cellules mésangiales Prolifération

30
GNMP La GNMP est une néphropathie glomérulaire devenue rare.
Plutôt les femmes. Le tableau clinique est soit celui d’un syndrome néphrotique impur, soit celui d’un syndrome néphritique

31
GNMP de type 1 Augmentation du nombre de cellules mésangiales
Expansion de la matrice mésangiale Empâtement diffus du floculus glomérulaire réalisant un aspect lobulaire Les parois capillaires glomérulaires sont épaissies En IF, on observe des dépôts granuleux de C3 dans le mésangium et dans les capillaires périphériques Un aspect en « rail de chemin de fer » peut être observé en coloration argentique ou avec le PAS.

34
GNMP de type 1 La GNMP de type I est rarement idiopathique. Un grand nombre d’affections infectieuses et auto-immunes peuvent être à l’origine de cette forme de GNMP Une baisse du complément (C3, C1q, C4) est fréquente et suggère une activation de la voie classique. Cette baisse du complément est fluctuante et peut être ignorée si les examens ne sont pas répétés.
 est fréquente et suggère une activation de la voie classique. Cette baisse du complément est fluctuante et peut être ignorée si les examens ne sont pas répétés.")
35
GNMP de type 1 Le pronostic de la GNMP de type 1 est mauvais
Les rémissions spontanées sont très rares Le traitement, basé sur les stéroïdes, est mal codifié et peu efficace Il doit être réservé aux patients ayant une protéinurie néphrotique, une insuffisance rénale et/ou une importante atteinte interstitielle sur la PBR Le traitement de la cause est important Le risque de récidive après transplantation est voisin de 90%

36
GNMP de type II La GNMP de type 2 se distingue par la présence de dépôts immuns denses à l’origine d’un épaississement d’aspect rubanné de la paroi capillaire glomérulaire en microscopie optique En microscopie électronique, il s’agit de dépôts électron-dense En IF sont retrouvés des dépôts de C3 et des différents autres composants du complément

39
GNMP de type II Même pronostic
La GNMP de type 2 peut s’accompagner d’une lipodystrophie partielle L’hypocomplémentémie est profonde, persistante et concerne la voie alterne du complément Il peut être mis en évidence la présence d’une immunoglobuline augmentant le catabolisme du C3 (C3 nephritic factor = C3Nef). Cet auto-anticorps se lie à la C3bBb (C3 convertase alterne) qui clive le C3 en C3a et C3b. Liée à cette Ig, la C3 convertase n’est plus dégradée et dégrade le C3 en continu. Même pronostic
. Cet auto-anticorps se lie à la C3bBb (C3 convertase alterne) qui clive le C3 en C3a et C3b. Liée à cette Ig, la C3 convertase n’est plus dégradée et dégrade le C3 en continu. Même pronostic.")
40
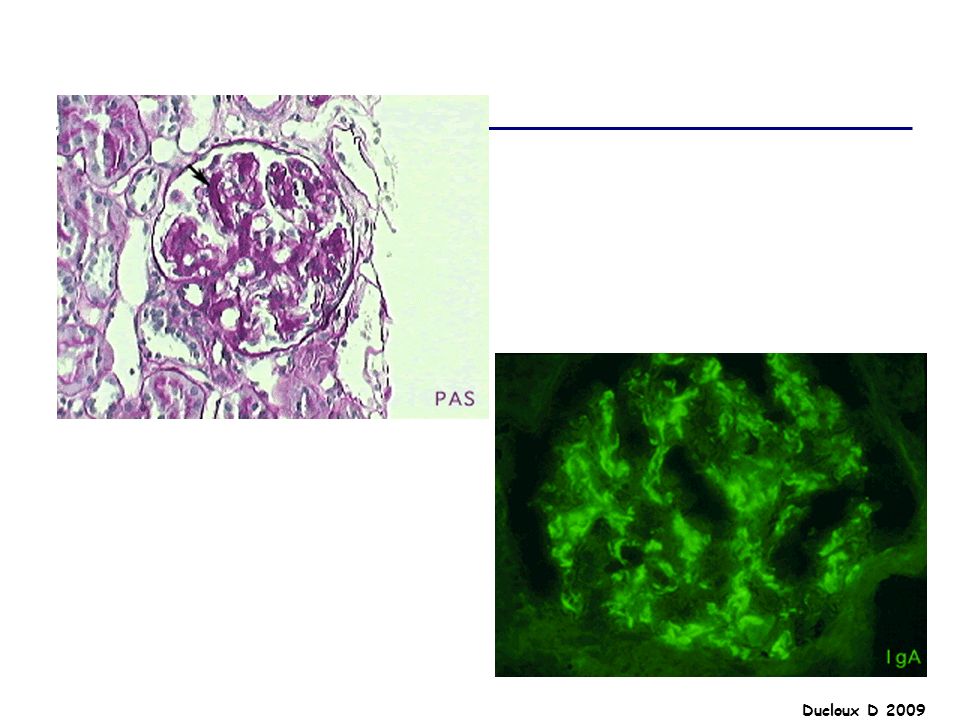
Néphropathie à IgA La néphropathie à IgA est la GN primitive la plus fréquente Il s'agit d'une GN à dépôts d'immuns complexes définie en immunohistologie par la présence exclusive ou prédominante de dépôts d'IgA accompagnée par des manifestations histologiques variables Il s'agit le plus souvent d'une maladie isolée et primitive, mais certaines formes peuvent être secondaires à d'autres pathologies

41
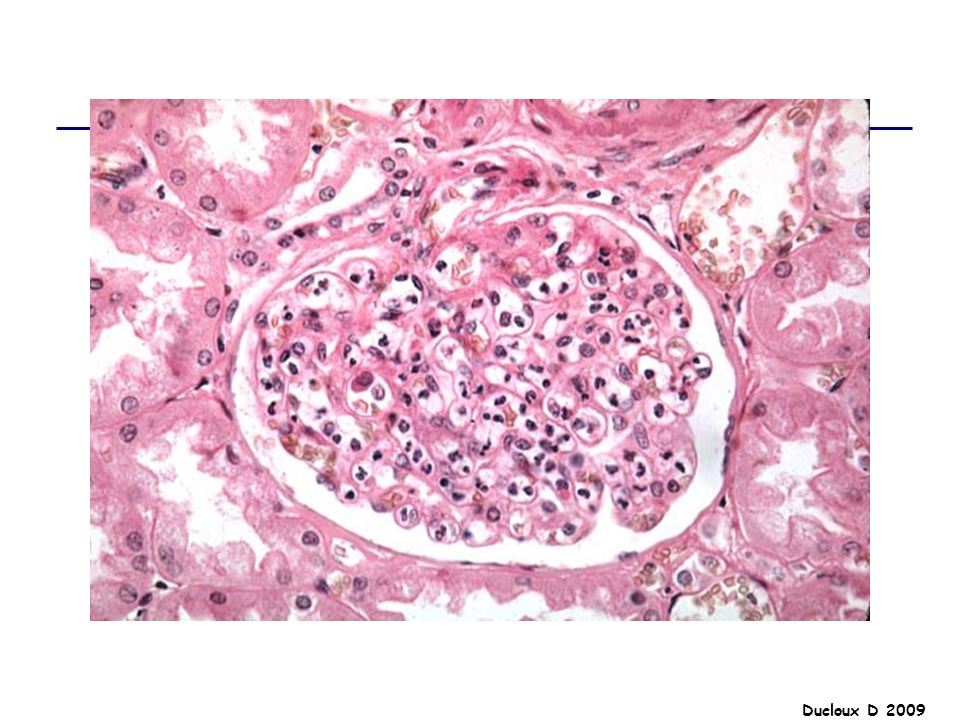
Prolifération diffuse
Proliféraion épithéliale

43
Néphropathie à IgA La néphropathie à IgA survient à tous les âges (maximum vers ans) Plus souvent chez l'homme que chez la femme Il existe des zones géographiques de plus forte incidence (Asie orientale) Des facteurs génétiques, mais aussi des différences de prise en charge diagnostique, peuvent expliquer ces différences Les sujets noirs sont plus rarement atteints
 Des facteurs génétiques, mais aussi des différences de prise en charge diagnostique, peuvent expliquer ces différences. Les sujets noirs sont plus rarement atteints.")
44
Néphropathie à IgA La ou les causes de la néphropathie à IgA sont inconnues Des facteurs génétiques et environnementaux intriqués expliquent probablement la survenue de cette maladie Une augmentation de synthèse des IgA et/ou un défaut de clairance de ces immunoglobulines, anomalies fréquemment observées dans la néphropathie à IgA ne peuvent suffire à expliquer les dépôts d'IgA dans le rein Les IgA sont des Ig glycosylées et il a été mis en évidence récemment un défaut de galactosylation des IgA chez les patients souffrant de néphropathie à IgA

45
Néphropathie à IgA Présentation clinique extrêmement protéiforme
Certains symptômes ou syndromes sont plus fréquents au cours de la néphropathie à IgA. une hématurie microscopique persistante associée à une protéinurie intermittente et modérée un syndrome des hématuries macroscopiques récidivantes, coïncidant souvent avec une infection ORL L'évolution est également très variable (15 à 40% des patients évolueraient vers l'IRC terminale) L'HTA, une score histologique élevé, une hématurie microscopique persistante, une protéinurie > 1g/j et une insuffisance rénale présente au moment du diagnostic sont des facteurs péjoratifs.
 L HTA, une score histologique élevé, une hématurie microscopique persistante, une protéinurie > 1g/j et une insuffisance rénale présente au moment du diagnostic sont des facteurs péjoratifs.")
46
Néphropathie à IgA Le traitement des néphropathies à IgA est mal codifié Basé sur des thérapeutiques non spécifiques (régime modérément restreint en protides, IEC). Dans les formes sévères, +/- l'adjonction d'immunosuppresseurs Une amygdalectomie est indiquée chez les patients ayant des angines à répétition accompagnées d'hématurie macroscopique Le risque de récurrence après transplantation est de 20 à 60%; néanmoins le plus souvent histologique ne compromettant que rarement la fonction du transplant (15% des récidives).
. Dans les formes sévères, +/- l adjonction d immunosuppresseurs. Une amygdalectomie est indiquée chez les patients ayant des angines à répétition accompagnées d hématurie macroscopique. Le risque de récurrence après transplantation est de 20 à 60%; néanmoins. le plus souvent histologique ne compromettant que rarement la fonction du transplant (15% des récidives).")
47
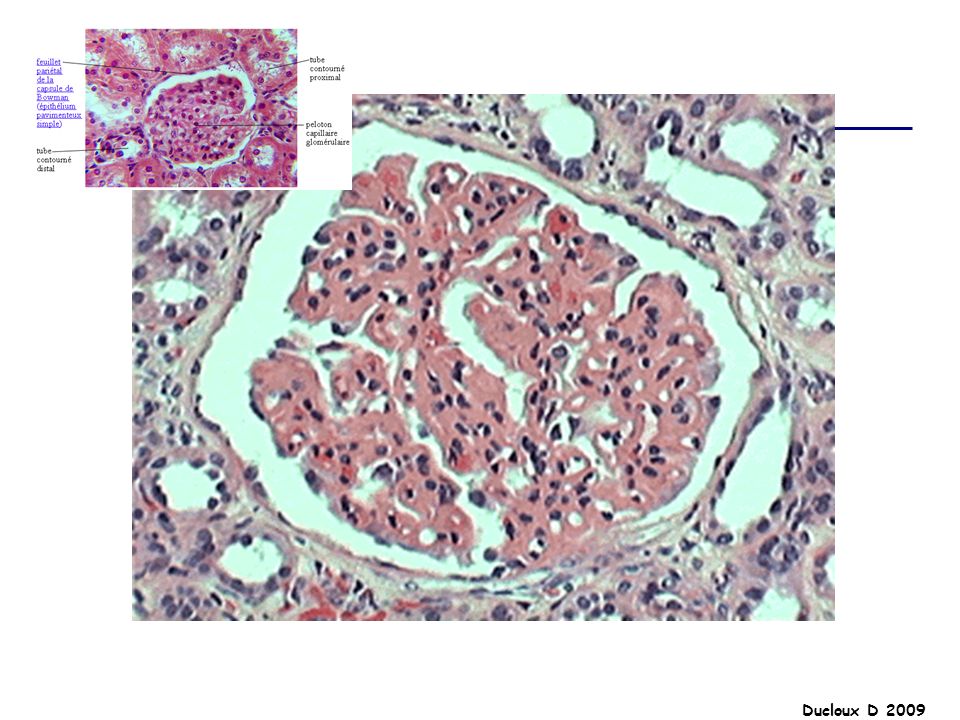
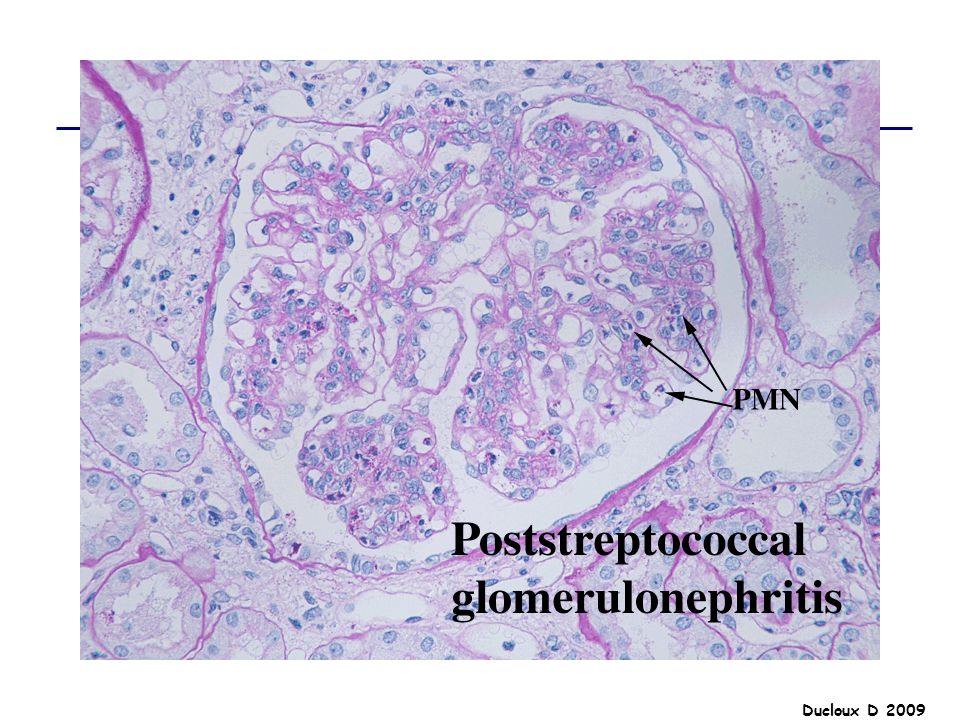
GNA La GNA post-streptococcique est la plus fréquente des GNA post-infectieuse enfants de moins de 10 ans, 5 à 14 jours après une infection pharyngée à streptocoque hémolytique du groupe A de type 12.. La présentation clinique se fait sous la forme d'un syndrome néphritique avec : Insuffisance rénale en règle modérée HTA souvent au premier plan avec risque d'encéphalopathie hypertensive Protéinurie sans syndrome néphrotique le plus souvent Hématurie macroscopique

48
GNA Le diagnostic peut être conforté par :
La présence d'anticorps antistreptococcique streptolysine O, DNAse, Hyaluronidase, NADase absente dans 1/3 des cas La diminution du CH50 constante à la phase aiguë retourne à la normale en environ 8 semaines

49
Prolifération diffuse
Proliféraion épithéliale Prolifération endocapillaire Afflux cellules inflammatoires

52
GNA L'évolution est en règle favorable
Les anomalies cliniques disparaissent en une à deux semaines et la fonction rénale se normalise en moins d'un mois L'hématurie microscopique et une proténurie modérée peuvent persister des mois, voire des années Des lésions rénales irréversibles sont observées chez moins de 1% des patients.

53
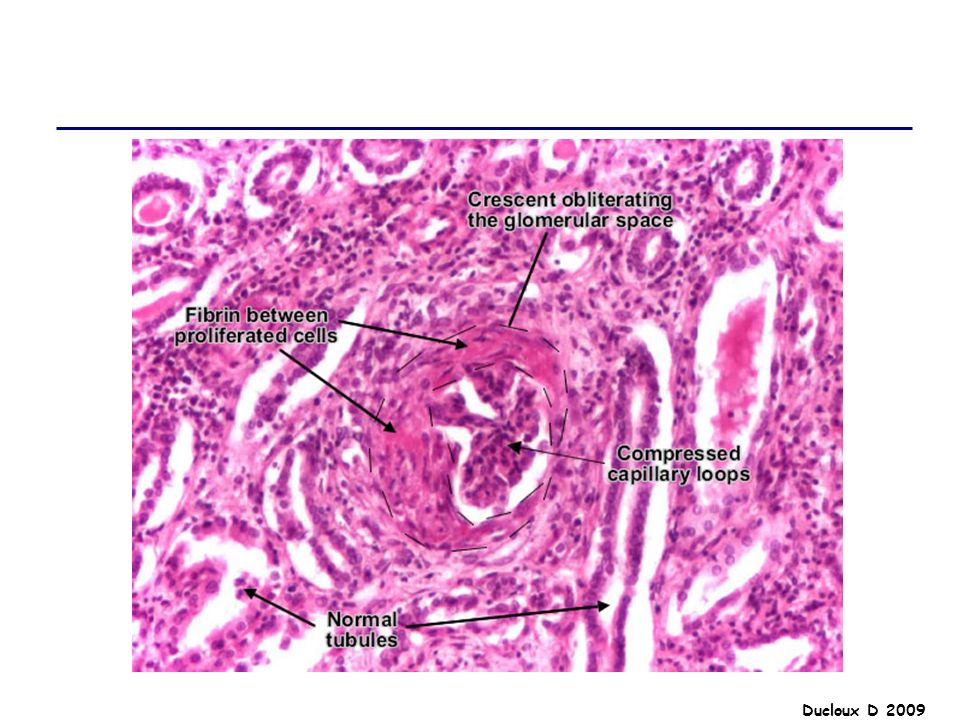
GNEC Prolifération Lésions tubulo- Formation de interstitielles
croissants Afflux cellules inflammatoires

56
GNEC Tableau clinique PBR Pronostic Etiologies Traitement
Dépôts granuleux MBG ANCA Traitement

57
GN secondaires

58
GN secondaires non prolifératives

59
Diabète GN la plus fréquente Diabète type 1 versus diabète type 2
Physiopathologie AGE Hyperfiltration Facteurs génétiques

61
Diabète

62
Diabète Diabète de type 1
Au début du diabète de type 1, il existe fréquemment une hyperfiltration glomérulaire et une augmentation de la taille des reins Des anomalies urinaires ne sont décelables qu’au bout de 10 ans d ‘évolution du diabète de type 1 (25% des patients) La première anomalie consiste en une petite augmentation de l’excrétion urinaire d’albumine (microalbuminurie). L’apparition d’une protéinurie se fait dans les 5 ans suivant celle de la microalbuminurie Avec la protéinurie surviennent une HTA et une détérioration progressive de la fonction rénale aboutissant à l’IRCt dans un délai moyen de 5 à 6 ans Les traitements récents ont modifiés l’histoire naturelle de la néphropathie du diabète de type 1 et permit de ralentir voire de stopper son évolution.
 La première anomalie consiste en une petite augmentation de l’excrétion urinaire d’albumine (microalbuminurie). L’apparition d’une protéinurie se fait dans les 5 ans suivant celle de la microalbuminurie. Avec la protéinurie surviennent une HTA et une détérioration progressive de la fonction rénale aboutissant à l’IRCt dans un délai moyen de 5 à 6 ans. Les traitements récents ont modifiés l’histoire naturelle de la néphropathie du diabète de type 1 et permit de ralentir voire de stopper son évolution.")
63
Diabète Diabète de type 2
L’histoire naturelle de la ND de type 2 est sensiblement différente : une microalbuminurie est fréquente au moment du diagnostic l’HTA est précoce et précède bien souvent les autres anomalies rénales la phase d’hyperfiltration glomérulaire est rarement identifiée Diagnostic plus difficile Indication PBR dans quelques cas

64
Diabète Traitement Le traitement (et la prévention) repose sur :
Le contrôle glycémique Le contrôle de la PA par les IEC ou les ARA2 Un objectif de 130/80 mm Hg est préconisé

65
Amylose Les amyloses sont définies par des dépôts extracellulaires de protéines ayant en commun des affinités tinctoriales, un aspect fibrillaire en microscopie électronique, et une conformation spatiale dite b-plissée Microscopie optique Coloration en rose par l’HES Biréfringence jaune-vert en lumière polarisée après coloration par le rouge Congo Fluorescence après coloration par la Thioflavine Microscopie électronique Fibrilles non organisées de 10 nm de et de longueur variable Diffraction aux rayons X Conformation en feuillets b-plissés perpendiculaires au grand axe de la fibrille

66
Amylose Des composants communs :
composant amyloïde P / glycosaminoglycans / apolipoprotéine E / autres substances (anti-protéases, …) Une protéine spécifique. Les différentes protéines spécifiques sont à la base de la classification des amyloses AL : La protéine est soit la chaîne légère d’Ig entière, soit une partie tronquée de cette chaîne légère contenant au moins le segment variable VL. AH : La protéine est un fragment de chaîne lourde gamma G1 AA : La protéine AA a un PM de 8 kDa constituée de 76 AA. Elle dérive de la protéine SAA.
 Une protéine spécifique. Les différentes protéines spécifiques sont à la base de la classification des amyloses. AL : La protéine est soit la chaîne légère d’Ig entière, soit une partie tronquée de cette chaîne légère contenant au moins le segment variable VL. AH : La protéine est un fragment de chaîne lourde gamma G1. AA : La protéine AA a un PM de 8 kDa constituée de 76 AA. Elle dérive de la protéine SAA.")
67
Amylose AL Complication des proliférations plasmocytaires monoclonales. La prolifération est soit patente, soit impossible à mettre en évidence (amylose primitive). pic de fréquence ~ 60 ans L’atteinte viscérale est souvent diffuse avec une prédilection pour le cœur, les reins, la peau, le tube digestif, le SNP, le canal carpien et l’os. Le composant monoclonal est soit une Ig monoclonale et/ou une chaîne légère d’Ig. La chaîne légère est plus souvent k que l Médiane de survie d’environ 12 mois. L’atteinte cardiaque et hépatique et l’association à un myélome sont de mauvais pronostic.
. pic de fréquence ~ 60 ans. L’atteinte viscérale est souvent diffuse avec une prédilection pour le cœur, les reins, la peau, le tube digestif, le SNP, le canal carpien et l’os. Le composant monoclonal est soit une Ig monoclonale et/ou une chaîne légère d’Ig. La chaîne légère est plus souvent k que l. Médiane de survie d’environ 12 mois. L’atteinte cardiaque et hépatique et l’association à un myélome sont de mauvais pronostic.")
68
Amylose AA L’amylose AA complique les maladies inflammatoires chroniques et plus particulièrement la PR Elle complique ces pathologies après une évolution longue (en moyenne 15 ans) Elle touche avec prédilection le rein, le foie, la rate, le tube digestif et moins souvent le cœur et les glandes endocrines L’insuffisance rénale est le principal facteur pronostique de la maladie Le pronostic est également mauvais avec une médiane de survie entre 2 et 4 ans
 Elle touche avec prédilection le rein, le foie, la rate, le tube digestif et moins souvent le cœur et les glandes endocrines. L’insuffisance rénale est le principal facteur pronostique de la maladie. Le pronostic est également mauvais avec une médiane de survie entre 2 et 4 ans.")
69
LUPUS L'atteinte rénale est fréquente au cours du LED et représente un aspect pronostic majeur de la maladie La moitié des patients ont des symptômes néphrologiques (protéinurie et/ou hématurie) au moment du diagnostic et plus des 3/4 en auront au cours de son évolution Il s'agit d'une complication précoce du LED, la plupart des patients développant une atteinte rénale dans les trois premières années suivant le diagnostic protéinurie (80% des patients) hématurie (40%) 1/3 des patients développent une insuffisance rénale
 au moment du diagnostic et plus des 3/4 en auront au cours de son évolution. Il s agit d une complication précoce du LED, la plupart des patients développant une atteinte rénale dans les trois premières années suivant le diagnostic. protéinurie (80% des patients) hématurie (40%) 1/3 des patients développent une insuffisance rénale.")
70
Classe I : Glomérules normaux
IA : strictement normaux IB : normaux en optique, mais dépôts en IF et ME Classe II : GN mésangiale IIA : épaississement de la matrice mésangiale et/ou hypercellularité modérée IIB : Hypercellularité plus importante Classe III : GN segmentaire et focale IIIA : Lésions nécrosantes actives IIIB : lésions sclérosantes et lésions actives IIIC : lésions sclérosantes Classe IV : GN proliférative diffuse IVA : sans lésion segmentaire IVB : avec lésions nécrosantes actives IVC : lésions sclérosantes et lésions actives IVD : lésions sclérosantes Classe V : GEM VA : pure VB : + classe II Classe VI : GN sclérosante

71
LUPUS Glomérulonéphrite mésangiale
atteinte la plus modérée parmi les lésions glomérulaires observées au cours du LED. Elle représente 10 à 20% des PBR de LED. Le tableau clinique regroupe le plus souvent : une protéinurie et une hématurie modérée rarement une HTA l'insuffisance et le syndrome néphrotique sont absents Elle survient en règle dans un contexte de maladie peu active (complément peu abaissé, faible augmentation des anticorps anti-DNA). Le pronostic est bon. Aucun traitement n'est indiqué.
. Le pronostic est bon. Aucun traitement n est indiqué.")
72
LUPUS Glomérulonéphrite proliférative focale
incidence voisine de la précédente. Le tableau clinique est plus sévère. protéinurie hématurie constantes / SN, HTA et insuffisance rénale 1/3 des patients. En microscopie optique : prolifération segmentaire < 50% des glomérules / nécrose rare. La prolifération intéresse principalement les cellules mésangiales et endothéliales. Les parois vasculaires sont soulignées par des dépôts éosinophiles donnant un aspect en anse de fil de fer (wire loop). D’autres aspects peuvent être observés (corps hématoxyliques de Gross : pathognomoniques de néphrite lupique) En IF, dépôts d'IgG et de C3 uniformes, diffus, épais (wire loop) ou fins (dépôts extramembraneux). [C1q et C4] Le pronostic de cette forme est difficile à préciser car les patients avec une atteinte atteignant moins de 20% des glomérules ont un excellent pronostic. Le traitement est discutable.
. D’autres aspects peuvent être observés (corps hématoxyliques de Gross : pathognomoniques de néphrite lupique) En IF, dépôts d IgG et de C3 uniformes, diffus, épais (wire loop) ou fins (dépôts extramembraneux). [C1q et C4] Le pronostic de cette forme est difficile à préciser car les patients avec une atteinte atteignant moins de 20% des glomérules ont un excellent pronostic. Le traitement est discutable.")
73
LUPUS Glomérulonéphrite proliférative diffuse
C'est la forme la plus sévère et la plus fréquente, représentant environ 50% des PBR de LED. L'HTA, l'insuffisance rénale et le syndrome néphrotique sont très fréquents, les anomalies du sédiment urinaire constantes. Il s'agit de formes cliniques de LED en règle très active avec hypocomplémentémie et élévation importante des anticorps anti-DNA. Les anomalies élémentaires sont identiques à celles des formes focales, mais plus étendues et plus sévères. Le pronostic en l'absence de traitement est mauvais et ces formes constituent en elles même une indication de traitement immunosuppresseur intensif

74
GNEC à ANCA Les GNEC avec ANCA regroupent trois vascularites atteignant les petits vaisseaux La maladie de Wegener La polyangéite microscopique La maladie de Churg et Strauss ANCA = anticorps anti-cytoplasme des polynucléaires) = famille d’auto-anticorps dirigés contre des antigènes du cytoplasme des polynucléaires neutrophiles (rôle pathogène direct) La technique de référence pour la mise en évidence des ANCA est lFI après fixation des PNN à l’éthanol. Deux aspects principaux peuvent être observés : Une fluorescence cytoplasmique (c-ANCA) WEGENER Une fluorescence périnucléaire (p-ANCA) La spécificité peut être déterminée en ELISA.
 = famille d’auto-anticorps dirigés contre des antigènes du cytoplasme des polynucléaires neutrophiles (rôle pathogène direct) La technique de référence pour la mise en évidence des ANCA est lFI après fixation des PNN à l’éthanol. Deux aspects principaux peuvent être observés : Une fluorescence cytoplasmique (c-ANCA) WEGENER. Une fluorescence périnucléaire (p-ANCA) La spécificité peut être déterminée en ELISA.")
75
Syndrome de Goodpasture
La maladie des anticorps anti-membrane basale est caractérisée par la présence d’anticorps circulants dirigés contre un antigène de la MBG et d’une atteinte rénale et/ou pulmonaire avec présence de ces anticorps détectée en immunofluorescence et réalisant un marquage linéaire des MBG et/ou des membranes alvéolaires Lorsqu’il existe une atteinte bipolaire, rénale et pulmonaire, on parle de syndrome de Goodpasture (SGP)
")
76
Syndrome de Goodpasture
Atteinte rénale : Il s’agit d’une GNRP Atteinte pulmonaire : L’hémorragie alvéolaire est le principal symptôme de l’atteinte pulmonaire du SGP. Elle peut se manifester par : Une hémoptysie : 1/3 des cas Une anémie ferriprive Des infiltrats pulmonaires sur la Rx Thorax Une augmentation de la diffusion du CO liée à la présence d’hémoglobine dans les alvéoles

77
Syndrome de Goodpasture
Devant une GNRP isolée ou associée à une atteinte rénale, le diagnostic de SGP doit être évoqué. Il est confirmé par : La présence d’anticorps circulants anti-MBG. Ces anticorps sont dirigés contre la partie NC1 de la chaîne alpha 3 du collagène de type IV. Ces anticorps peuvent être détectés par IF indirecte (40% de faux négatifs) ou mieux par méthode ELISA (5% de faux négatifs) qui permet en outre de mesurer et suivre le taux d’anticorps. La PBR qui met en évidence une GNEC avec des dépôts linéaires d’IgG le long des MBG et parfois des tubules.
 ou mieux par méthode ELISA (5% de faux négatifs) qui permet en outre de mesurer et suivre le taux d’anticorps. La PBR qui met en évidence une GNEC avec des dépôts linéaires d’IgG le long des MBG et parfois des tubules.")
79
Syndrome d’Alport Génétique
La maladie est transmise selon un mode lié à l’X Chez la femme, 50% environ des cellules sont atteintes (théorie de l’inactivation aléatoire de l’X) et les symptômes sont modérés ou absents Le gène muté sur le chromosome X code pour la chaîne alpha 5 du collagène 4 (COL4A5). Les mutations survenant sur la chaîne alpha 5 modifie la structure de la MBG qui ne contient pas de chaînes alpha 3, 4 et 5, mais garde des chaînes alpha 1 et 2, caractéristique de la vie fœtale et plus sensibles aux attaques protéolytiques par les collagénases et les cathepsines. La sévérité des manifestations cliniques semble dépendre du type de mutation. Plus rarement, la transmission est autosomique récessive. Le gène concerné est situé sur le chromosome 2 et code pour les chaînes alpha 3 et alpha 4. Les symptômes sont identiques à ceux de la forme liée à l’X.
 et les symptômes sont modérés ou absents. Le gène muté sur le chromosome X code pour la chaîne alpha 5 du collagène 4 (COL4A5). Les mutations survenant sur la chaîne alpha 5 modifie la structure de la MBG qui ne contient pas de chaînes alpha 3, 4 et 5, mais garde des chaînes alpha 1 et 2, caractéristique de la vie fœtale et plus sensibles aux attaques protéolytiques par les collagénases et les cathepsines. La sévérité des manifestations cliniques semble dépendre du type de mutation. Plus rarement, la transmission est autosomique récessive. Le gène concerné est situé sur le chromosome 2 et code pour les chaînes alpha 3 et alpha 4. Les symptômes sont identiques à ceux de la forme liée à l’X.")
80
Syndrome d’Alport Rénales
La symptomatologie rénale débute chez H vers l’âge de 5 ans protéinurie et une hématurie microscopique avec parfois des épisodes d’hématurie macroscopique Une HTA et une insuffisance rénale apparaissent plus tard. L’âge moyen de l’IRC terminale est variable, en règle entre 15 et 40 ans. En microscopie électronique simple amincissement de la MBG Progressivement apparaissent des zones de fragmentation longitudinale de la MBG responsables d’un aspect laminé et feuilleté. Les anomalies observées en optique sont moins spécifiques (hypercellularité focale du glomérule, infiltrat interstitiel avec lipophages).
.")
81
Syndrome d’Alport Extra-rénales Surdité de perception
Lenticône antérieur, cataracte, lésions périmaculaires Thrombopathie avec plaquettes géantes (formes autosomiques récessives) Leiomyosarcomes
 Leiomyosarcomes.")
82
Syndrome d’Alport Les éléments du diagnostic comprennent :
Histoires clinique et familiale PBR Biopsie cutanée avec marquage par anticorps anti-chaîne alpha 5 Diagnostic génétique

Présentations similaires












>")










