Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Cours de Pharmacologie générale Pr.SLIMANI.M mslimani20@yahoo.fr

2
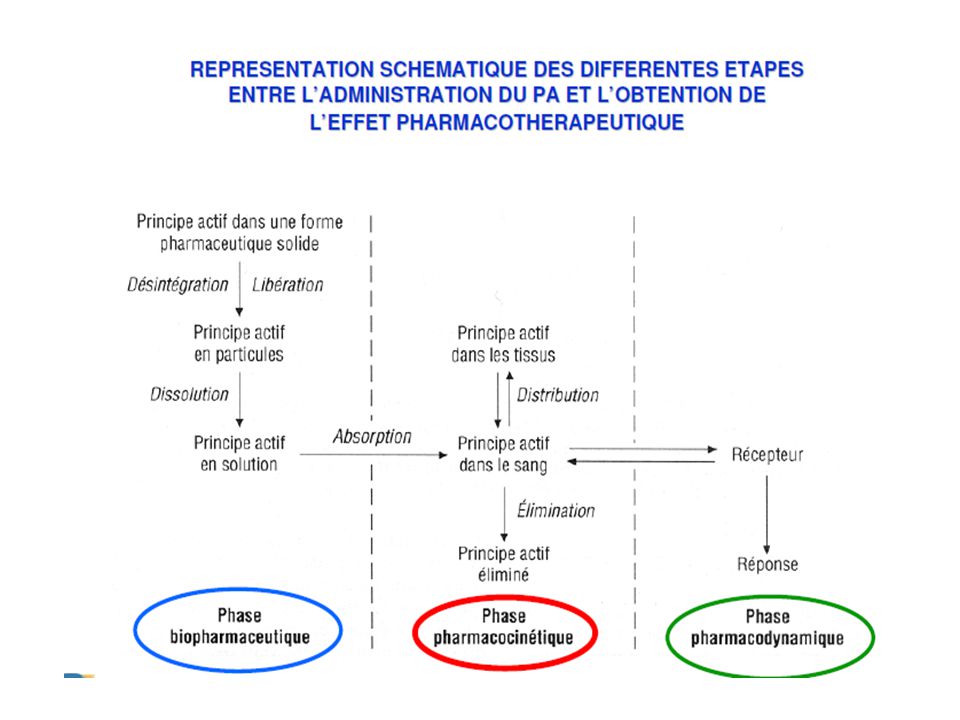
La pharmacologie Pharmacodynamie Pharmacocinétique
(pharmacology) Vient du mot grec “ Pharmakon ” qui veut dire remède mais aussi poison. Discipline ayant pour objet l’étude des interactions entre les médicaments et les organismes vivants. Pharmacodynamie Pharmacocinétique Action du médicament sur l’organisme Action de l’organisme sur le médicament
 Vient du mot grec Pharmakon qui veut dire remède mais aussi poison. Discipline ayant pour objet l’étude des interactions entre les médicaments et les organismes vivants. Pharmacodynamie. Pharmacocinétique. Action du médicament sur l’organisme. Action de l’organisme sur le médicament.")
3
la pharmacovigilance et la pharmacodépendance, l’étude du médicament chez l’animal (pharmacologie expérimentale), ou chez l’homme (pharmacologie clinique). La pharmacologie moléculaire étudie les propriétés physico-chimique des médicaments et leur relation avec leur activité biologique. L’étude de l’utilisation des médicaments, de leur contexte et de ses conséquences pour la société est l’objet de la pharmacoépidémiologie, de la pharmacoéconomie et de la pharmacologie sociale La plupart des principes actifs actuels sont préparés par synthèse chimique intégrale ou par semi synthèse à partir de substances naturelles. Excipients La présence d’excipients est indispensable pour assurer la conservation du médicament et jouent aussi un rôle important dans la vitesse de mise à disposition de l’organisme du principe actif

4
Les excipients sont classés selon leur fonction en :
- agrégants : excipients qui assurent la cohésion d’un mélange de poudres et permettent la réalisation de comprimés - diluants ou véhicules : phase continue qui permet la solution ou la dispersion des constituants du médicament dans un volume suffisant - intermèdes : substances permettant la réalisation physique du médicament ou assurant sa stabilité (par exemple, émulsionnant) - colorants : substances colorées servant de témoin d’homogénéité d’un mélange de poudres ou à identifier le médicament fini - édulcorants ou correctifs : modificateurs du goût permettant de rendre une préparation agréable ou de masquer le mauvais goût d’un principe actif - conservateurs : substances destinées à empêcher la dégradation chimique ou l’altération microbiologique d’un médicament.
 - colorants : substances colorées servant de témoin d’homogénéité d’un mélange de poudres ou à identifier le médicament fini. - édulcorants ou correctifs : modificateurs du goût permettant de rendre une préparation agréable ou de masquer le mauvais goût d’un principe actif. - conservateurs : substances destinées à empêcher la dégradation chimique ou l’altération microbiologique d’un médicament.")
6
Les étapes de la genèse d’un effet
Principe actif administré Bactéries Insectes Parasites ABSORPTION Interactions Cibles pharmacologiques Concentrations Plasma Concentrations Biophase DISTRIBUTION ELIMINATION Action cellulaire Réponse fonctionnelle PHARMACOCINETIQUE PHARMACODYNAMIE

7
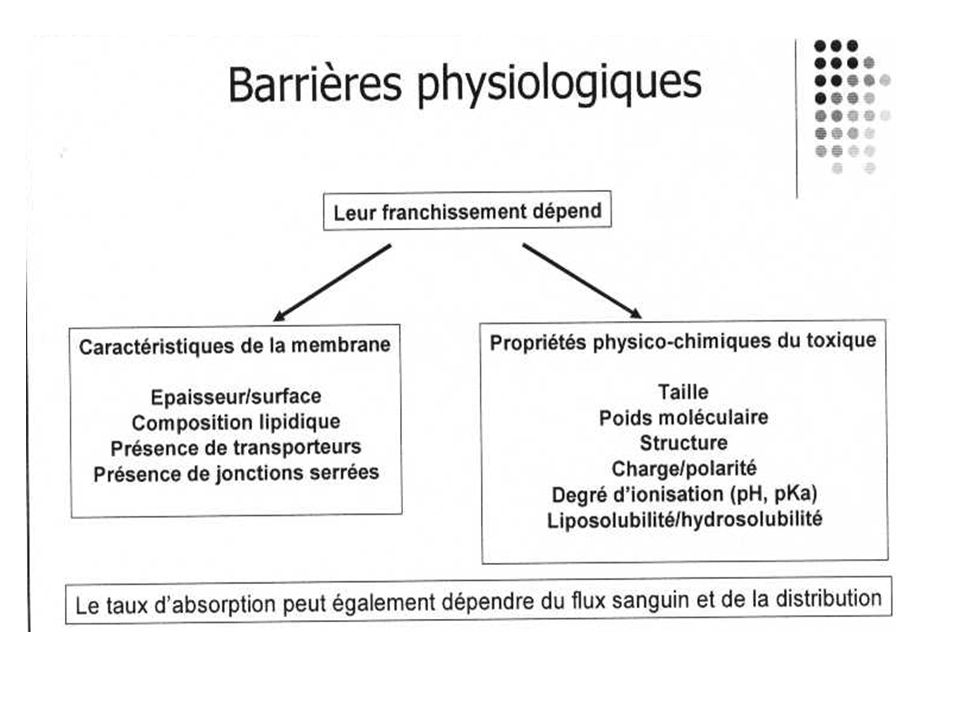
La vitesse et la durée de l'absorption dépendent de :
- l'état physique du médicament et de la libération du principe actif : cette mise à disposition constitue la « phase galénique ». L'ordre décroissant de vitesse de passage est : solutions aqueuses > solutions huileuses > suspensions > solides - la concentration : plus elle est forte et plus le passage est rapide - la circulation : plus le tissu est vascularisé et plus le passage est rapide (muscle). Les tissus peu vascularisés retiennent les médicaments (graisse). La vasodilatation et la vasoconstriction accélèrent ou ralentissent l'absorption ; on peut les provoquer dans ce but - la surface : plus la surface d'absorption est grande, plus le passage est rapide et important.
. Les tissus peu vascularisés retiennent les médicaments (graisse). La vasodilatation et la vasoconstriction accélèrent ou ralentissent l absorption ; on peut les provoquer dans ce but. - la surface : plus la surface d absorption est grande, plus le passage est rapide et important.")
8
Médicament (drug, medicine, medication)
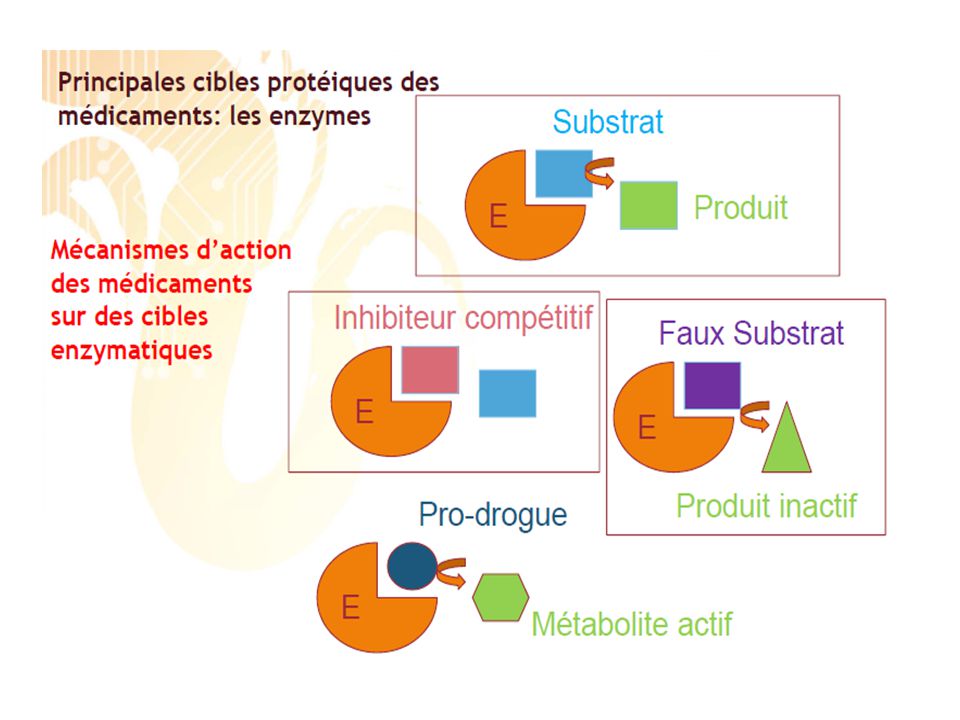
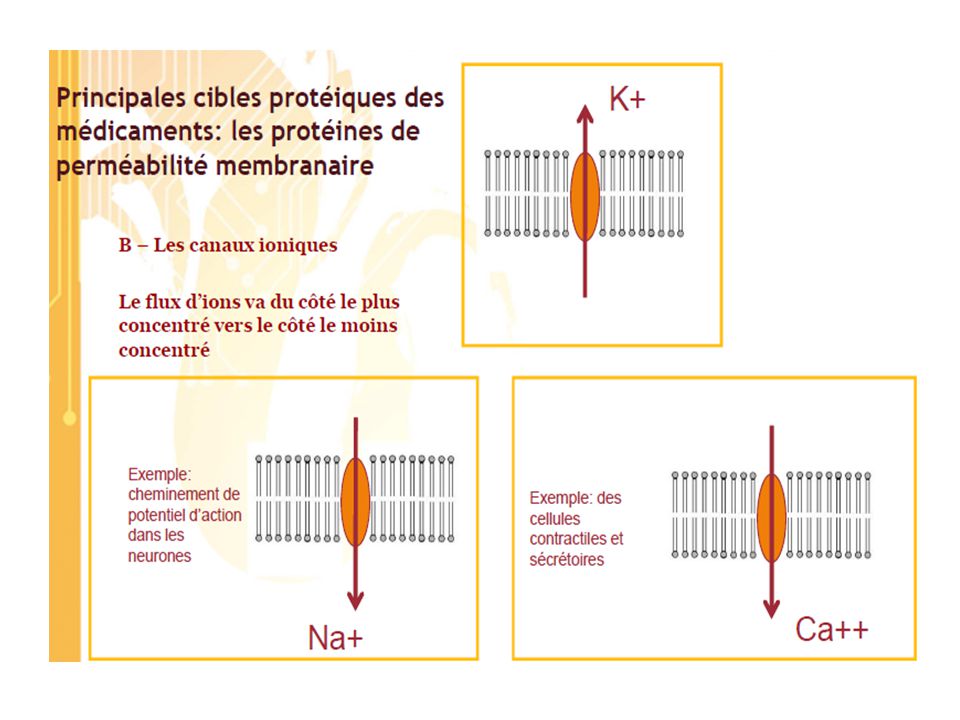
“ Toute substance ou composition, présentée comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales, ainsi que tout produit administré à l’homme ou à l’animal en vue d’établir un diagnostic médical, de restaurer, corriger ou modifier leurs fonctions organiques Un médicament est défini par : •Une DCI = Dénomination Commune Internationale Principe actif •Un nom chimique •Un nom commercial 1 Caractéristiques des médicaments et mécanisme d’action moléculaire 1.1 Cibles des médicaments Les cibles des médicaments actuels sont peu nombreuses par rapport à la multiplicité des cibles potentielles issues des connaissances récentes. En effet, aujourd’hui, on note l’existence d’environ 3000 substances utilisées comme médicaments mais uniquement 500 cibles. Les deux classes de cibles les plus importantes sont les récepteurs membranaires des médiateurs (45% des cibles actuelles) et les enzymes (28%). Les canaux ioniques (5%) et les récepteurs nucléaires (2%) sont d’autres cibles potentielles des médicaments
 et les enzymes (28%). Les canaux ioniques (5%) et les récepteurs nucléaires (2%) sont d’autres cibles potentielles des médicaments.")
9
Comment agit un médicament sur la cible ?
- La rencontre entre le médicament et la cible Pour qu’un médicament engendre un effet thérapeutique, c’est-à-dire pour qu’il soit efficace, il faut tout d’abord qu’il y ait rencontre entre ce médicament et sa cible. Pour que cette mise en contact soit possible, le médicament doit être présent en quantité suffisante au niveau de cette cible. Ceci dépend de la dose administrée, mais aussi de sa capacité à atteindre sa cible : il ne doit pas être dégradé lorsqu’il circule dans le sang, il doit pouvoir franchir certaines barrières… Enfin, la rencontre entre le médicament et la cible dépend de l’affinité et de la sélectivité du médicament. Ces deux dernières contraintes sont liées à la forme et à la structure électronique des deux molécules. En effet, pour se rencontrer, médicament et cible doivent avoir une complémentarité structurale (c’est-à-dire tridimensionnelle) et électronique. La première est facilitée par la flexibilité des molécules, c’est-à-dire par leur capacité à changer de conformation (de forme) lorsque le médicament s’approche de la cible : on parle de conformation induite. La complémentarité électrostatique caractérise les interactions entre charges de signe opposé. Lorsque ces deux complémentarités existent, l’affinité entre le médicament et sa cible est très grande
 et électronique. La première est facilitée par la flexibilité des molécules, c’est-à-dire par leur capacité à changer de conformation (de forme) lorsque le médicament s’approche de la cible : on parle de conformation induite. La complémentarité électrostatique caractérise. les interactions entre charges de signe opposé. Lorsque ces deux complémentarités existent, l’affinité entre le médicament et sa cible est très grande.")
10
Lorsqu’un médicament atteint une cible, une liaison va s’établir
-La liaison formée peut être une liaison forte, appelée liaison covalente mais il s’agit généralement d’une interaction faible. Le nombre de médicaments qui établissent directement des liaisons covalentes avec leur récepteur est assez restreint: l’aspirine.

11
La liaison covalente est une interaction forte, irréversible, qui résulte de la mise en commun par deux atomes de deux électrons qui les lient. L’établissement et la rupture des liaisons covalentes jouent un rôle capital dans le métabolisme, la biotransformation des médicaments et les effets qu’ils initient. -La liaison non covalente (liaison ionique, liaison hydrophobe, liaison hydrogène…) tient compte des effets d’attraction et de répulsion. C’est une liaison fragile. Cependant, un médicament génère généralement un grand nombre de liaisons non covalentes avec la cible ce qui favorise une bonne interaction et stabilise le complexe formé.
 tient compte des effets d’attraction et de répulsion. C’est une liaison fragile. Cependant, un médicament génère généralement un grand nombre de liaisons non covalentes avec la cible ce qui favorise une bonne interaction et stabilise le complexe formé.")
12
Conséquences des interactions médicament/cible:
La liaison d’un médicament à un récepteur est l’étape nécessaire à une suite de réponses. Elle peut induire : · Un changement de conformation du récepteur ( canaux ioniques ) : ceci modifie l’activité du récepteur · Des modifications de la distribution des charges électroniques, des échanges de protons… · L’activation de protéines, ce qui conduit à une cascade de modifications : o Production des nouvelles substances, appelées messagers intracellulaires, qui initient des voies de signalisation du message o Régulation de facteurs de transcription
 : ceci modifie l’activité du récepteur. · Des modifications de la distribution des charges électroniques, des échanges de protons… · L’activation de protéines, ce qui conduit à une cascade de modifications : o Production des nouvelles substances, appelées messagers intracellulaires, qui initient des voies de signalisation du message. o Régulation de facteurs de transcription.")
13
Pro médicaments: ou précurseurs « prodrugs » , sont des dérivés devant subir une biotransformation enzymatique avant d’exercer leur effet pharmacologique exemple : pro médicament: Talampicilline Médicament :Ampicilline ester phtalidylique de ampicilline peu lipobhile, 40 % absorbés diarrhées (par lipophile , 70% absorbés, perturbation de la flore intestinale pour être pas d’effet intrinsèque antibactérien absorbé Pro médicament Dérivé dépourvu des propriétés pharmacologiques Médicament Composé possédant les propriétés pharmacologiques

14
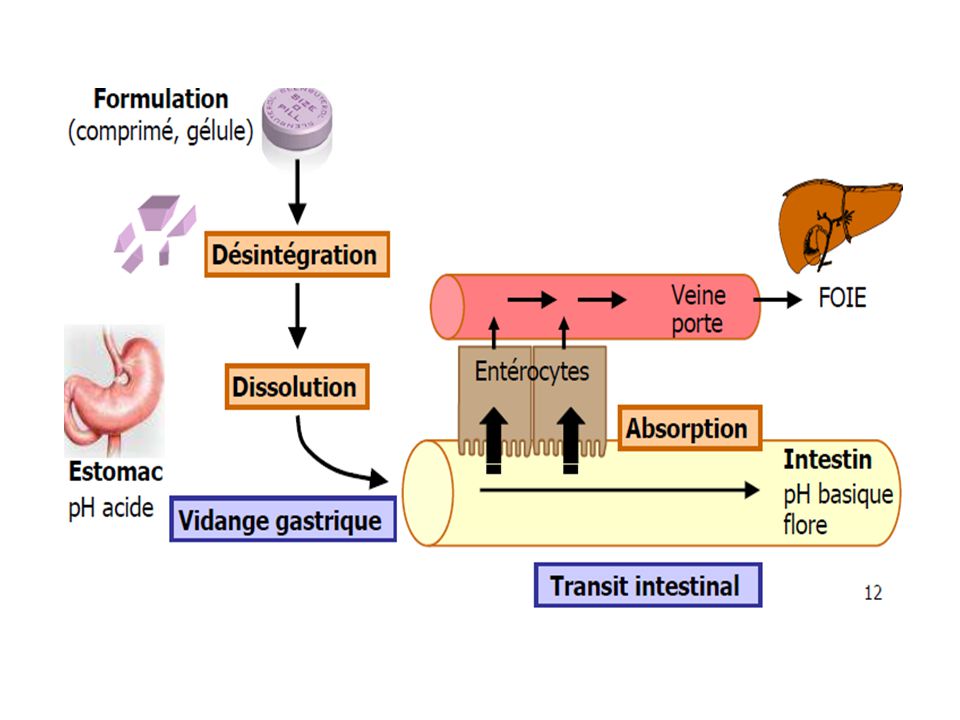
La phase biopharmaceutique constitue la mise à disposition de l’organisme des principes actifs. Elle comprend une étape de libération, qui a généralement lieu par désagrégation de la forme solide en particules de petite taille, suivie d’une étape de dissolution, qui consiste en une dispersion d’un principe actif à l’état moléculaire en milieu aqueux, au site d’absorption. Le temps de désagrégation constitue l’un des essais à effectuer après la fabrication de comprimés ou de capsules

17
Comprimés Les comprimés sont fabriqués par compression d’un mélange de poudre constitué du principe actif et d’excipients.. Les comprimés non enrobés doivent se désagréger en moins de 15 minutes. Pour les comprimés enrobés, le temps de désagrégation dépend de la nature de l’enrobage. Ce dernier peut être constitué de substances diverses : résines, gommes, sucres, colorants, etc. En fonction du type d’enrobage, on distingue Les objectifs d’un enrobage peuvent être : - masquage d’une odeur ou d’un goût désagréable - protection du principe actif, contre la lumière ou l’humidité

18
Formes à libération accélérée
Les comprimés à libération accélérée sont formulés de façon à obtenir un temps de désagrégation court. Ils comprennent : - comprimés effervescents comprimés orodispersibles :faciliter la prise du médicament, en cas de problème de déglutition par exemple

19
Formes orales : Solides : comprimés , gélules , capsules Liquides : sirops, solutions , suspensions Les gélules: formées de 2 demi-capsules à base de gélatine, emboîtées et contenant un mélange de poudre sèche Il existe des gélules : - gastro-résistantes (enveloppe ou contenu enrobés) - à libération modifiée avantages : masque saveur désagréable protection du PA
 - à libération modifiée. avantages : masque saveur désagréable. protection du PA.")
20
Les sirops - forte concentration en sucre - administration facile et saveur agréable / enfants - mais dosage imprécis… c. à café = 5 ml c. à dessert = 10 ml c. à soupe = 15 ml Les solutions ou solutés buvables - PA dans un solvant à base d’eau, ou eau + alcool -Présentation en ampoules (unitaire) ou en flacon Solutions injectables : Médicaments déposé à l’intérieur des tissus ou dans le torrent circulatoire Différentes présentations Solutions : IM, IV, SC, ID Suspensions : IM, SC, ID Emulsions : IV Formes à libération prolongée : IM ou SC
 ou en flacon. Solutions injectables : Médicaments déposé à l’intérieur des tissus ou dans le torrent circulatoire. Différentes présentations. Solutions : IM, IV, SC, ID. Suspensions : IM, SC, ID. Emulsions : IV. Formes à libération prolongée : IM ou SC.")
21
mouvements entre les compartiments
Ext. Système Nerveux Central volume extra- cellulaire compartiment central : volume qui est en équilibre d’échange rapide avec le plasma = essentiellement le liquide extracellulaire

24
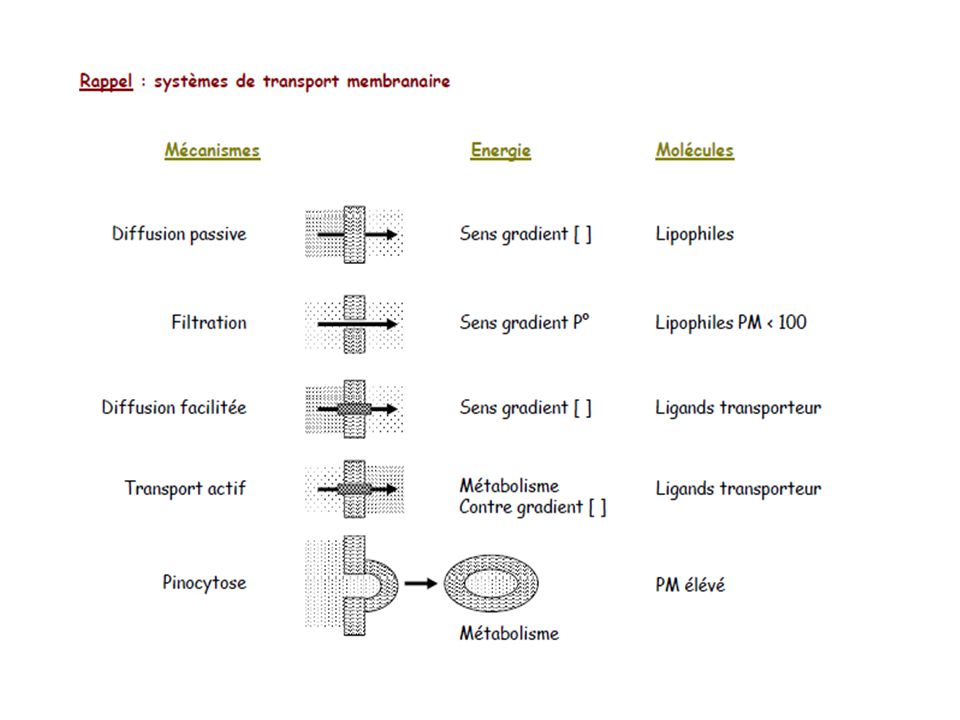
Toxico-cinétique: principes de base
Diffusion à travers les membranes la vitesse de diffusion dépend de la surface d’absorption S du coefficient de perméabilité (Kp) du gradient des concentrations de part et d’autre de la membrane C C2 flux net = Kp.S.(C2-C1) Kp: coefficient de perméabilité Kp = D . k1 / e k1 : coefficient de partage huile/eau e : épaisseur de la membrane (env. 50 Ă pour les membranes biologiques) D : coefficient de diffusion dans les lipides seules les petites molécules non chargées et peu polaires passent facilement à travers les membranes
 du gradient des concentrations de part et d’autre de la membrane. C1 C2. flux net = Kp.S.(C2-C1) Kp: coefficient de perméabilité. Kp = D . k1 / e. k1 : coefficient de partage huile/eau. e : épaisseur de la membrane (env. 50 Ă pour les membranes biologiques) D : coefficient de diffusion dans les lipides. seules les petites molécules non chargées et peu polaires. passent facilement à travers les membranes.")
27
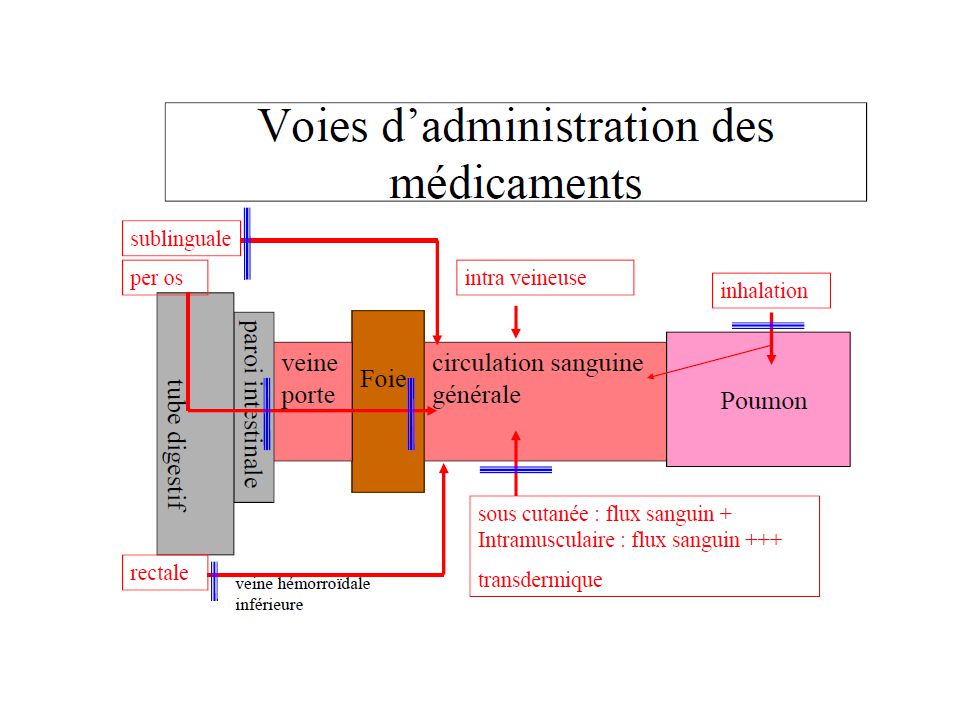
Administration orale, perlinguale, rectal
Les voies d’administration: Voie entérale Administration orale, perlinguale, rectal Le PA est absorbé au niveau buccale, de l’estomac, de l’intestin grêle ou du rectum. Voie parentérale Administration IV, IM, SC. Le PA est injecté à travers la peau à l’aide d’un dispositif médical stérile (aiguille + seringue/ cathéter/ microperfuseur)… Voie topique, transcutané Le PA agit directement sur la muqueuse (nasale, respiratoire,…) ou est appliqué directement sur la peau Autres voies (oculaire, nasale, auriculaire)
… Voie topique, transcutané. Le PA agit directement sur la muqueuse (nasale, respiratoire,…) ou est appliqué directement sur la peau. Autres voies (oculaire, nasale, auriculaire)")
29
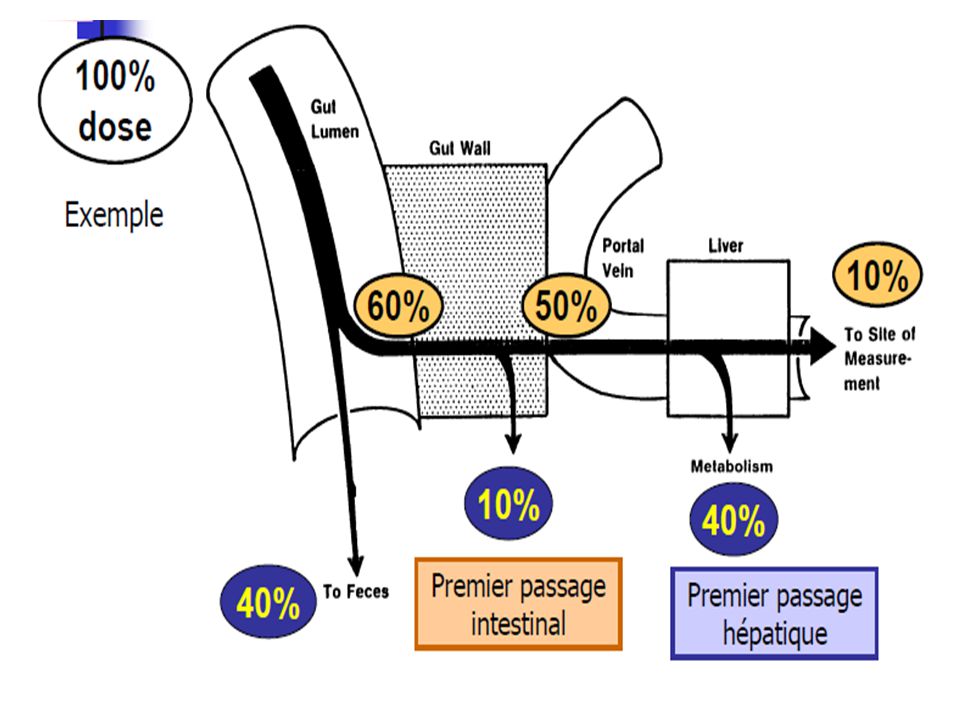
Phénomènes limitant la biodisponibilité pour l’administration par voie orale
paroi intestinale foie veine porte parvient dans le compartiment central Métabolisme hépatique Métabolisme Intra-intestinal non réabsorbé ou détruit, élimination fécale

30
Trajet d'un médicament administré par voie buccale

31
Voie orale ou per os Le tube digestif va de la bouche au rectum. Les membranes que le médicament doit franchir sont l'épithélium digestif et l'endothélium vasculaire. La voie orale n'est pas utilisable si le médicament destiné à un traitement général est dégradé dans le tube digestif (pH, flore microbienne, enzymes du tube digestif) L'absorption digestive peut se faire à tous les niveaux du tube digestif. Bouche : L'absorption du médicament par la muqueuse buccale qui permet une absorption rapide et évite le passage hépatique est généralement appelée voie perlinguale. .
 L absorption digestive peut se faire à tous les niveaux du tube digestif. Bouche : L absorption du médicament par la muqueuse buccale qui permet une absorption rapide et évite le passage hépatique est généralement appelée voie perlinguale. .")
32
Estomac : les substances actives acides faibles sont absorbées au niveau de l'estomac, car elles ne sont pas ionisées en milieu acide ; sous forme non-ionisée liposoluble, elles sont par conséquent capables de franchir les barrières biologiques lipophiles. Les substances actives bases faibles sont elles faiblement absorbées au niveau de l'estomac, car elles sont ionisées en milieu acide

33
En cas de différence de pH entre les deux côtés de la barrière lipidique, le passage du médicament est favorisé dans le sens du milieu acide vers le milieu alcalin pour un acide faible et en sens inverse pour une base faible

34
La surface de l'estomac est d'environ 1m2
La surface de l'estomac est d'environ 1m2. Le pH du liquide gastrique est acide. Le débit de drainage sanguin de l'estomac est faible, environ 0,2 L/min. Sont absorbées au niveau de l'estomac les molécules neutres et les acides non ionisés à pH acide. Sont sécrétées dans le liquide gastrique à partir du sang de nombreuses molécules, notamment les bases qui s'ionisent par protonation en arrivant dans le liquide gastrique acide, selon la réaction :

35
La transformation intra gastrique :
Certaines substances sont instables en milieu acide. Sur le plan pharmacocinétique, l'absorption digestive de la pénicilline G est médiocre, inférieure à 30%, car elle est détruite par le suc gastrique acide (rapidement hydrolysé).C’est ce qui explique qu’il est préférable de lui substituer la Pénicilline V lorsqu’on désir e employer par la voie orale, étant donné que leurs spectre antibactérien sont semblables La lévodopa est métabolisée dans l’estomac par une décarboxylase. Les principaux métabolites sont des acides phénylcarboxyliques et de la méthoxydopa. Lorsque l’on retarde la vidange gastrique , on augmente la décomposition intra gastrique et on diminue par conséquent la biodisponibilité
.C’est ce qui explique qu’il est préférable de lui substituer la Pénicilline V lorsqu’on désir e employer par la voie orale, étant donné que leurs spectre antibactérien sont semblables. La lévodopa est métabolisée dans l’estomac par une décarboxylase. Les principaux métabolites sont des acides phénylcarboxyliques et de la méthoxydopa. Lorsque l’on retarde la vidange gastrique , on augmente la décomposition intra gastrique et on diminue par conséquent la biodisponibilité.")
36
L’acidité gastrique n’a pas que des effets négatifs
L’acidité gastrique n’a pas que des effets négatifs. Ainsi, il a été démontré qu’elle était nécessaire à la transformation du chlorazépate en nordiazépam. La chlorazépate est très polaire , très peu liposoluble et par conséquent très peu absorbé au niveau gastro-intestinal. Il est donc nécessaire qu’il se transforme en nordiazépam pour être absorbé Intestin : absorption au niveau de l’intestin grêle (milieu plutôt basique). Les substances actives acides faibles sont faiblement absorbées au niveau de l'intestin grêle, car elles sont ionisées en milieu basique ; sous forme ionisée hydrosoluble, elles franchissent difficilement les barrières biologiques lipophiles. Les substances actives bases faibles, elles, sont bien absorbées à ce niveau ! La surface de l'intestin est grande : 200 à 300m2. Le pH est alcalin : 6 à 8. L'irrigation sanguine est importante, 1 L/minute. La majorité des médicaments sont absorbés à ce niveau
. Les substances actives acides faibles sont faiblement absorbées au niveau de l intestin grêle, car elles sont ionisées en milieu basique ; sous forme ionisée hydrosoluble, elles franchissent difficilement les barrières biologiques lipophiles. Les substances actives bases faibles, elles, sont bien absorbées à ce niveau ! La surface de l intestin est grande : 200 à 300m2. Le pH est alcalin : 6 à 8. L irrigation sanguine est importante, 1 L/minute. La majorité des médicaments sont absorbés à ce niveau.")
37
Les médicaments peuvent la traverser par trois mécanismes :
- la muqueuse intestinale se conduit comme une barrière lipidique ; le pH à sa surface est différent de celui du contenu intestinal et égal à 5,3. Les médicaments liposolubles dont le pK est compris entre 3 et 8 (acides et bases faibles) traversent la barrière, à la différence des corps hydrosolubles ou ionisés A l’inverse, les cellules intestinales peuvent constituer un barrage actif à la pénétration dans l’organisme de certains médicaments bien qu’ils aient franchi la membrane cellulaire
 traversent la barrière, à la différence des corps hydrosolubles ou ionisés. A l’inverse, les cellules intestinales peuvent constituer un barrage actif à la pénétration dans l’organisme de certains médicaments bien qu’ils aient franchi la membrane cellulaire.")
38
Deux mécanismes sont concernés :
- les cellules intestinales peuvent transformer (métaboliser) en général partiellement, certains médicaments. Elles disposent pour cela d’enzymes microsomales, comme le cytochrome CYP3A4 le système de la P-glycoprotéine constitue un mécanisme de transport actif capable de rejeter dans la lumière intestinale soit le principe actif lui-même, soit ses produits de dégradation. Ceci explique la faible biodisponibilité de certaines substances lipophiles Une particularité de l'absorption digestive est le métabolisme de premier passage: Le médicament, absorbé au niveau du tube digestif, passe par le foie, atteint le coeur et après passage pulmonaire se distribue dans l'ensemble de l'organisme. Au niveau de la muqueuse intestinale et du foie, le médicament rencontre des enzymes susceptibles de le transformer en un ou plusieurs métabolites parfois actifs mais le plus souvent inactifs. C'est le métabolisme de premier passage (First pass metabolism) qui explique la moindre efficacité de certains médicaments, surtout lorsqu'ils sont administrés à doses faibles, car ils sont en grande partie métabolisés avant d'arriver dans le sang
 en général partiellement, certains médicaments. Elles disposent pour cela d’enzymes microsomales, comme le cytochrome CYP3A4. le système de la P-glycoprotéine constitue un mécanisme de transport actif capable de rejeter dans la lumière intestinale soit le principe actif lui-même, soit ses produits de dégradation. Ceci explique la faible biodisponibilité de certaines substances lipophiles. Une particularité de l absorption digestive est le métabolisme de premier passage: Le médicament, absorbé au niveau du tube digestif, passe par le foie, atteint le coeur et après passage pulmonaire se distribue dans l ensemble de l organisme. Au niveau de la muqueuse intestinale et du foie, le médicament rencontre des enzymes susceptibles de le transformer en un ou plusieurs métabolites parfois actifs mais le plus souvent inactifs. C est le métabolisme de premier passage (First pass metabolism) qui explique la moindre efficacité de certains médicaments, surtout lorsqu ils sont administrés à doses faibles, car ils sont en grande partie métabolisés avant d arriver dans le sang.")
40
Voie rectale : la surface absorbante totale varie entre 200 et 400 cm2 , par rapport à celle du petit intestin : cm2.l’absorption se fait par diffusion passive. La destruction de la forme galénique est fonction de la nature de l’excipient: un excipient fondant dans le rectum (matière grasses comme le beurre de cacao , ou glycérides semi synthétiques), leur point de fusion sera un facteur essentiel et celui doit être compris entre ° et 37.6°, l’optimum se situant à 36.5° Un excipients hydrosolubles (masse gélatine/glycérine ou polyoxyéthyèneglycols ), la vitesse de destruction est proportionnelle à la vitesse de dissolution de l’excipient dans le liquide du rectum. Quelque soit l’excipient utilisé , après fusion ou dissolution , une masse plus ou moins visqueuse qui formera un film à la surface de la muqueuse , film à partir duquel le P.A va effectuer son transfert vers le liquide rectal. Les veines hémorroidales inférieurs et moyennes aboutissent aux veines iliaques internes qui se jettent dans la veine cave inférieure, évitant ainsi le premier passage hépatique , soustrait ainsi aux effets des sucs gastriques ( env.30% ). Les veines hémorroidales supérieures sont reliées à la veine mésentérique inférieure qui mène le sang à la veine porte puis au foie.
, leur point de fusion sera un facteur essentiel et celui doit être compris entre 32.6 ° et 37.6°, l’optimum se situant à 36.5° Un excipients hydrosolubles (masse gélatine/glycérine ou polyoxyéthyèneglycols ), la vitesse de destruction est proportionnelle à la vitesse de dissolution de l’excipient dans le liquide du rectum. Quelque soit l’excipient utilisé , après fusion ou dissolution , une masse plus ou moins visqueuse qui formera un film à la surface de la muqueuse , film à partir duquel le P.A va effectuer son transfert vers le liquide rectal. Les veines hémorroidales inférieurs et moyennes aboutissent aux veines iliaques internes qui se jettent dans la veine cave inférieure, évitant ainsi le premier passage hépatique , soustrait ainsi aux effets des sucs gastriques ( env.30% ). Les veines hémorroidales supérieures sont reliées à la veine mésentérique inférieure qui mène le sang à la veine porte puis au foie.")
41
La perlinguale, sublinguale (sous la langue) - absorption très facile par cette voie cette absorption se fait au niveau de la muqueuse buccale par diffusion passive, elle permet une résorption rapide en limitant l'effet de premier passage hépatique (au niveau du foie) puisque la résorption se fait par les veines jugulaires qui se jettent directement dans la veine cave supérieure. On a donc une action très rapide et une action maximale du médicament La voie pulmonaire L'absorption se fait par inhalation. on peut utiliser des substances fragiles qui seraient détruites ou altérées au niveau digestif les poumons ont une surface alvéolaire importante, qu'ils sont dotés d'un débit sanguin élevée et que les échanges entre l'air alvéolaire et le sang sont intenses - des concentrations plasmatiques et tissulaires élevées pour certains produits action des médicaments directement au niveau pulmonaire

43
L'absorption est d'autant plus rapide que le gaz est soluble dans le sang. Les particules atmosphériques quant à elles sont différemment absorbées et éliminées en fonction de leur dimension.

44
La voie cutanée La pénétration se fait au niveau de l'épiderme, et elle dépend de différents facteurs :- l'état de la peau, si elle est lésée (plaie, brûlure, eczéma), le passage est plus important - l'âge du sujet (passage plus important chez les jeunes du à la teneur en eau plus importante) -la nature de l'excipient (certains excipients sont dépourvus de pouvoir de pénétration comme la vaseline) .On les réserve donc à une action strictement locale, par contre il en existe d'autres qui vont manifester un pouvoir pénétrant qui va permettre un certain passage dans le milieu intérieur La première barrière rencontré est l'épiderme et surtout la couche cornée ; de petites quantités de substances polaires peuvent la traverser ;les substances non polaires y diffusent grâce à leur liposolubilté. Le derme, barrière moins sélective ,est plus facilement franchissable La voie oculaire On a par cette voie une pénétration par diffusion à travers la cornée et la conjonctive
, le passage est plus important. - l âge du sujet (passage plus important chez les jeunes du à la teneur en eau plus importante) -la nature de l excipient (certains excipients sont dépourvus de pouvoir de pénétration comme la vaseline) .On les réserve donc à une action strictement locale, par contre il en existe d autres qui vont manifester un pouvoir pénétrant qui va permettre un certain passage dans le milieu intérieur. La première barrière rencontré est l épiderme et surtout la couche cornée ; de petites quantités de substances polaires peuvent la traverser ;les substances non polaires y diffusent grâce à leur liposolubilté. Le derme, barrière moins sélective ,est plus facilement franchissable. La voie oculaire. On a par cette voie une pénétration par diffusion à travers la cornée et la conjonctive.")
45
La voie intramusculaire
localisation : cadran fesse quart supérieur externe .La résorption est variable pour une même substance en fonction de la richesse de vascularisation du muscle Cette administration est réalisée sous forme d’un dépôt que le P.A doit abandonner afin d’atteindre le flux sanguin ou circulation lymphatique par pénétration ou perméation. Les facteurs susceptibles de modifier cette absorption : -ce qui sont propre à l’organisme receveur : âge du sujet , la taille : le poids du sujet , la température corporelle( T°: de la durée d’action et T° : de la vitesse d’absorption ), flux sanguin ( 0.02 à0.07 ml/min/g), coef.partage, pH milieu , nature du solvant utilisé , volume et concentration de la solution injectée) Facteurs relatifs à la forme galénique: qu’il s’agit d’une solution aqueuse ou huileuse , d’une suspension ou d’une émulsion, la libération du P.A actif dépend de nombreux facteurs : forme chimique (acide , base , sel , éther ),concentration du P.A dans le véhicule ,volume du l’absorption liquide injecté ,type de solvant (aqueux , organique, huileux), vitesse de dissolution du P.A dans le dépôt, taille des particules du P.A dans les suspensions ,coef de partage, présence d’agents favorisant l’absorption (hyaluronidase) , présence d’agents vasoconstricteurs .
, flux sanguin ( 0.02 à0.07 ml/min/g), coef.partage, pH milieu , nature du solvant utilisé , volume et concentration de la solution injectée) Facteurs relatifs à la forme galénique: qu’il s’agit d’une solution aqueuse ou huileuse , d’une suspension ou d’une émulsion, la libération du P.A actif dépend de nombreux facteurs : forme chimique (acide , base , sel , éther ),concentration du P.A dans le véhicule ,volume du l’absorption liquide injecté ,type de solvant (aqueux , organique, huileux), vitesse de dissolution du P.A dans le dépôt, taille des particules du P.A dans les suspensions ,coef de partage, présence d’agents favorisant l’absorption (hyaluronidase) , présence d’agents vasoconstricteurs. .")
46
La voie intraveineuse localisation : pli du coude main ,pied .Cette voie est utilisée comme voie d'urgence, mais également pour des perfusions en continu. Les intraveineuses se font toujours lentement. avantages : - biodisponibilité maximale (100%) on peut injecter des médicaments qui seraient irritants pour le tube digestif perfusion intraveineuse : permet de délivrer une quantité de PA par unité de temps: concentration stable, administration de fortes quantités de PA sans dépasser un certain seuil de concentration
 on peut injecter des médicaments qui seraient irritants pour le tube digestif. perfusion intraveineuse : permet de délivrer une quantité de PA par unité de temps: concentration stable, administration de fortes quantités de PA sans dépasser un certain seuil de concentration.")
47
Le volume apparent de distribution (Vd) est le volume fictif, exprimé en litres ou en litres par kilogramme, dans lequel se serait distribué la substance toxique en supposant que sa concentration soit homogène, c'est-à-dire que la concentration tissulaire moyenne soit identique à celle du plasma. On a Vd = dose / C0 (concentration initiale) La demi-vie plasmatique d'une substance toxique (T½) est le temps nécessaire pour que la concentration plasmatique diminue de moitié, par exemple de 100 à 50 mg/l. Elle est calculée à partir de l’inverse de la pente de décroissance des concentrations plasmatiques en substance au cours du temps.
 La demi-vie plasmatique d une substance toxique (T½) est le temps nécessaire pour que la concentration plasmatique diminue de moitié, par exemple de 100 à 50 mg/l. Elle est calculée à partir de l’inverse de la pente de décroissance des concentrations plasmatiques en substance au cours du temps.")
48
Cinétique lors d’une administration unique (per os par exemple)
10 20 4 8 12 16 temps [h] lC] (ou [Q]) A = compartiment d’administration A Q = Vd . [C] élimination
 A = compartiment d’administration. A. Q = Vd . [C] élimination.")
49
Mesure du volume de distribution (Vd) et de la constante d’élimination (ke)
Vd = Q / Ci C = Ci*exp(-ke.t) Log(C)=Ci - ke.t ke = pente de la droite injection i.v. de la quantité Q au temps 0. Ci
 Log(C)=Ci - ke.t. ke = pente de la droite. injection i.v. de la quantité Q. au temps 0. Ci.")
50
la clairance :correspond au volume de sang totalement épuré de la substance toxique par unité de temps. Elles est donc généralement exprimé en l/h. Plus la clairance est élevé, plus les capacités d’élimination de la substance par l’organisme sont importante. Cette notion de clairance prend donc en compte tous les processus d’élimination qu’ils s’agissent d’élimination sous forme inchangée (rénale…) et des différentes biotransformations (intestinale, hépatique…). On peut donc parler de notion de clairance (Cl) totale et de clairances hépatique, intestinale ou rénale, la première étant la résultante de toutes les autres : Cl totale = Cl rénale + Cl hépatique +… -La clairance et la demi-vie sont reliées par la notion de volume de distribution (Vd), paramètre pharmacocinétique de distribution: CL = Vd / T1/2 On conçoit que la clairance totale dépend de la constante d'élimination et donc de la T½ et du Vd. La clairance est une constante en cinétique linéaire. C = C0.exp -Kel T
, paramètre pharmacocinétique de distribution: CL = Vd / T1/2. On conçoit que la clairance totale dépend de la constante d élimination et donc de la T½ et du Vd. La clairance est une constante en cinétique linéaire. C = C0.exp -Kel T.")
51
L’évolution des concentrations au cours du temps est la résultante de l’entrée du médicament dans l’organisme et de son élimination. A la différence de l’administration i.v. unique, les processus d’absorption et d’élimination coexistent et l’aspect de la courbe variera avec les durées respectives de chacune de ces phases :

52
Calcul de la surface sous la courbe (SSC) par la méthode des trapèzes
On distingue : La biodisponibilité absolue, correspondant au rapport de la quantité absorbée par une voie d’administration donnée à celle obtenue par voie i.v. (égale à 100%, par définition). La biodisponibilité relative, permettant de comparer entre elles deux formes du médicament administrées par la même voie (ex. comprimé vs sirop). La comparaison porte alors sur les 3 paramètres : F, Cmax et Tmax.. Calcul de la surface sous la courbe (SSC) par la méthode des trapèzes
. La biodisponibilité relative, permettant de comparer entre elles deux formes du médicament administrées par la même voie (ex. comprimé vs sirop). La comparaison porte alors sur les 3 paramètres : F, Cmax et Tmax.. Calcul de la surface sous la courbe (SSC) par la méthode des trapèzes.")
53
Biodisponibilité par voie orale de 3 médicaments
. Mode de calcul de la biodisponibilité Eventuellement corrigée du rapport des doses administrées si celles-ci sont différentes :F(%) = [Dose i.v. x SSCforme étudiée / Dose forme étudiée x SSCi.v.] x 100 Biodisponibilité par voie orale de 3 médicaments
 = [Dose i.v. x SSCforme étudiée / Dose forme étudiée x SSCi.v.] x 100. Biodisponibilité par voie orale de 3 médicaments.")
54
Rapport efficacité/toxicité en fonction de la dose
Comme, d'une manière générale, l'augmentation des doses augmente à la fois l'efficacité et la toxicité, il faut tenir compte du rapport efficacité/toxicité en fonction de la dose. Cette courbe montre qu'il existe une dose optimum.

57
Schéma simplifié de l'oxydation d'un médicament par le cytochrome P-450

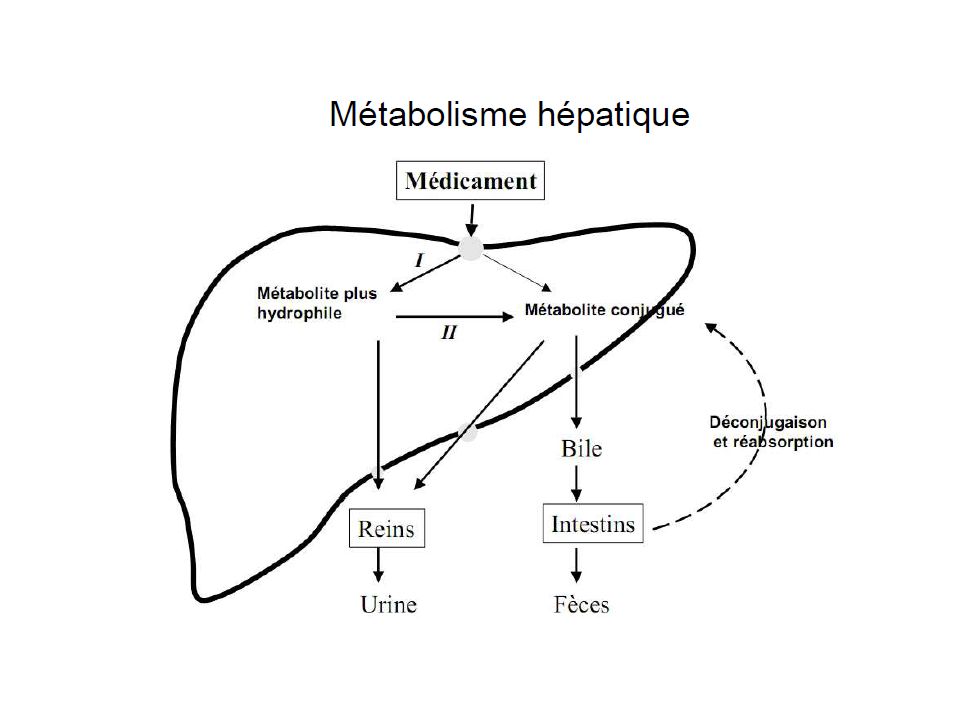
61
Réactions de phase II utilisation d’un groupe fonctionnel pour former une liaison covalente avec une molécule fortement hydrosoluble. En général, enzymes cytoplasmiques conjugaison avec l’acide glucuronique conjugaison avec un sulfate acétylation (à partir d’acétyl-Co-A) conjugaison avec une cystéine ou un glutathion (tripeptide avec une cystéine) ou autre acides aminé, glycine, glutamine, taurine méthylation (à partir du donneur S-adénosylmethionine (par ex. réaction d’inactivation des catécholamines par la COMT
 conjugaison avec une cystéine ou un glutathion (tripeptide avec une cystéine) ou autre acides aminé, glycine, glutamine, taurine. méthylation (à partir du donneur S-adénosylmethionine (par ex. réaction d’inactivation des catécholamines par la COMT.")
62
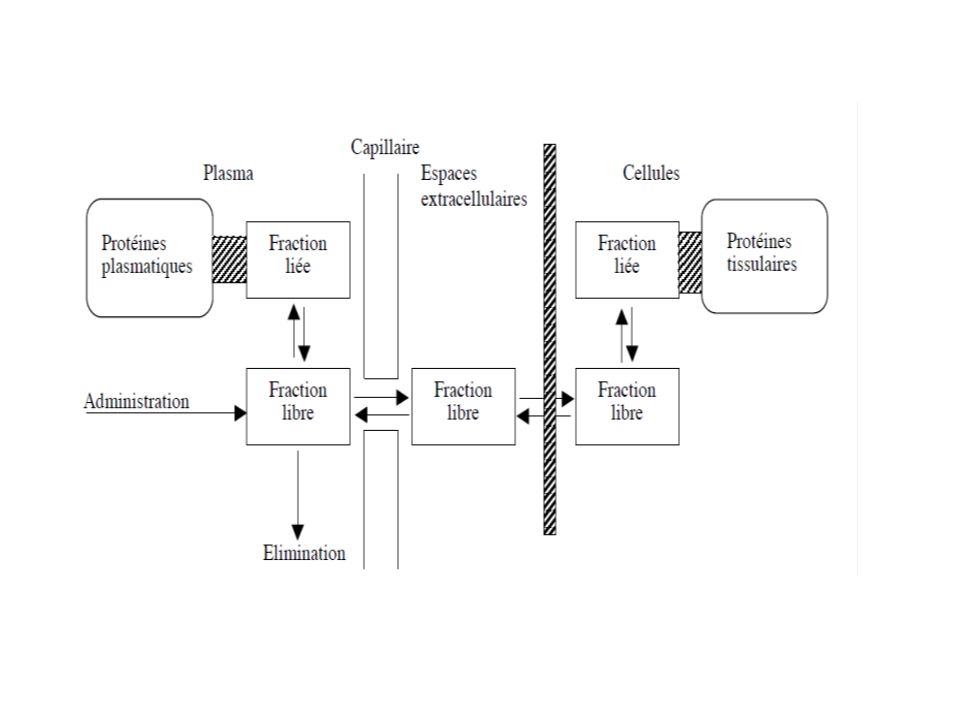
Transport sanguin Le sang joue le rôle d’un véhicule de transport par les hématies et les protéines circulantes susceptibles de fixer la substance médicamenteuse. On parle alors de fixation aux protéines plasmatiques. Cette fixation est réversible. La substance médicamenteuse se retrouve alors sous forme libre ou liée aux protéines. Notons ici que seul le médicament libre est pharmacologiquement actif : on perçoit alors toute l’importance de cette étape de distribution dans le devenir du médicament dans l’organisme. Quelles sont les protéines concernées ? Peut-on définir l’importance de la liaison aux protéines ? Quelles sont les conséquences de la liaison ? La fraction libre peut-elle varier ?

64
FIXATION DES MEDICAMENTS SUR LES PROTEINES PLASMATIQUES

65
La fixation est définie par le pourcentage de liaison pouvant aller de 0 à 100%. Par exemple, la fixation aux protéines plasmatiques de certains anti-inflammatoires est de 95%. La fixation du paracétamol est par contre nulle. On considère qu’une substance est fortement liée si son pourcentage de fixation dépasse 75%. –Soit [P] la concentration molaire de la protéine, [M] la concentration molaire du médicament, et [MP] la concentration de la liaison protéine-médicament ; on écrit la liaison de la façon suivante, afin d’exprimer la réversibilité : [P] + [M] [MP] Il y a équilibre entre la forme libre [M] et la forme liée [PM]. Si [M] augmente, [MP] augmente aussi. Si on diminue [M], alors [MP] va diminuer et un rééquilibre va être atteint entre la forme libre et la forme liée. Cette liaison est fondamentale, car seul le médicament sous sa forme libre est actif [M]. Aussi, la forme libre est diffusible à travers les membranes, et peut être éliminée et/ou métabolisée . La forme liée agit comme une réserve qui ne traverse pas les membranes

66
Fixation au niveau des éléments figurés
La fixation aux éléments figurés apparaît moins grande que celle relative aux protéines plasmatiques. Il est important de souligné ici l’interaction entre les médicaments et les hématies. Cette fixation est importante par exemple avec le propranolol ou le phénobarbital. Les phénomènes de déplacement semblent différents à ceux observés avec les protéines. Ce type de fixation est à considérer dès lors que le volume de distribution est faible et que la concentration érythrocytaire est forte comparée à la concentration au niveau plasmatique, car on prend en compte les concentrations au niveau plasmatique, pas au niveau sanguin.

67
Les facteurs limitant de la diffusion tissulaire sont : - la fixation aux protéines tissulaires qui déterminera la forme libre - les caractéristiques physico-chimiques de la molécule, à savoir sa masse molaire, la lipophilie (coefficient de partage), le pKa de la molécule (ou sa ionisation ou non ionisation) et donc de sa capacité à franchir les membranes vasculaires et cellulaires - de l’irrigation des organes et du débit sanguin : L’obstacle membranaire n’existe pas pour les substances de faible masse molaire et les composés liposolubles. la distribution dans les différents organes est variable, du fait des différents débits sanguins des tissus concernés Il faut distinguer les organes bien irrigués, comme le foie, les reins, le cœur, les poumons et le cerveau, et les organes ou tissus peu perfusés comme l’os, la peau et les graisses. Il existe une corrélation entre le vitesse de perfusion tissulaire et la vitesse de distribution vers ce même tissu

68
pénétration dans le système nerveux central (SNC) et le liquide céphalo-rachidien (LCR)
- une première barrière qui sépare le plasma du cerveau : barrière hémato encéphalique - une barrière située entre le plasma et le LCR : barrière hémato méningée - barrière qui sépare la substance nerveuse du LCR : barrière méningo encéphalique Ces barrières empêchent en principe la pénétration des médicaments liés aux protéines, des médicaments hydrosolubles et des ions. Par contre les médicaments liposolubles seront absorbés passage des médicaments à travers le placenta (échanges foeto-maternels). Le placenta n'empêche pas le passage d'un médicament du compartiment maternel vers le compartiment foetal, il peut seulement le limiter. Le passage placentaire d'un médicament obéit aux mêmes règles que pour les autres membranes
. Le placenta n empêche pas le passage d un médicament du compartiment maternel vers le compartiment foetal, il peut seulement le limiter. Le passage placentaire d un médicament obéit aux mêmes règles que pour les autres membranes.")
69
Sur le plan pharmacologique, ce type d’interférence se traduit par l’augmentation de la fraction libre plasmatique de l’un ou des deux médicaments présents. Il en résulte une augmentation des intensités des effets observés . Les principales substances ionisées susceptibles d’entrer en compétition au niveau des sites albuminiques . L’interaction la plus classique est celle de la warfarine (Anticoagulant )associée à la phénylbutazone . (anti-inflammatoire )Chez un sujet traité par l’antivitamine K à dose efficace, l’administration de phénylbutazone provoque sa défixation partielle, majorant l’effet anticoagulant. Aux concentrations thérapeutiques de ces deux substances, le pourcentage de forme libre plasmatique de warfarine passe de 10 à 30 p. cent … Il en résulte une augmentation importante des concentrations tissulaires de warfarine, celles-ci étant sensiblement trois fois plus élevées. Au niveau du foie où se trouvent les récepteurs de la warfarine, l’effet anticoagulant est multiplié par trois, ce qui, compte tenu du mauvais coefficient chimiothérapeurique de cette substance, se traduit par un surdosage générateur d’hémorragies. »
associée à la phénylbutazone . (anti-inflammatoire )Chez un sujet traité par l’antivitamine K à dose efficace, l’administration de phénylbutazone provoque sa défixation partielle, majorant l’effet anticoagulant. Aux concentrations thérapeutiques de ces deux substances, le pourcentage de forme libre plasmatique de warfarine passe de 10 à 30 p. cent … Il en résulte une augmentation importante des concentrations tissulaires de warfarine, celles-ci étant sensiblement trois fois plus élevées. Au niveau du foie où se trouvent les récepteurs de la warfarine, l’effet anticoagulant est multiplié par trois, ce qui, compte tenu du mauvais coefficient chimiothérapeurique de cette substance, se traduit par un surdosage générateur d’hémorragies. ».")
Présentations similaires
























cinétique/pharmaco (toxico)dynamie>")





